New bio-amphiphilic structures of sucrose via amides linkages
Abstract
A series of four new derivatives of sucrose have been synthesized using the straightforward methodology in order to give a mono substituted analogs of sucrose at C-6’ of fructose moiety. The synthesis was started from the reaction of sucrose with tert-butylchlorodiphenylsilane, which is able to react with an only less steric hindrance hydroxyl group at C-6’ due to its bulky structure. The other hydroxyl groups were acetylated by the reaction with acetic anhydride in pyridine. Then free the hydroxyl group at C-6’ again by the treatment with t-butylammonium fluoride in THF. The later was activated by conversion to a good leaving group via tosylation, followed by functionalized via azidation to give the precursor of the target series hepta-O-acetyl-6’-azido- sucrose. The precursor was coupled with four alkyl halides (C12, C8-4, C14, C10-8) via Staudinger reaction to produced the target structure after deacetylating. The purity and chemical structure of the synthesized compound was confirmed by CHN elemental analysis, high-resolution mass and 1H, 13C NMR spectroscopy.
Keywords
Staudinger reaction, Zemplen reaction, Sucrose core, silylation, biosurfactant
Introduction
Sucrose is a natural nonreducing disaccharide consist of two monosaccharide glucose and fructose in the form of (β-D-fructofuranosyl-α-D-glucopyranoside). The main sources of sucrose are sugar beet or sugar cane, which grows in many countries all over the world. Sucrose is the cheapest biodegradable carbohydrates, which produced in an industrial scale and this production is excess the need of the population. The eight alcoholic groups in its structure increased the complexity, sensitivity and functionality of sucrose. The availability, biodegradability, solubility and functionality make sucrose great starting material for numerous of transformational reactions achieved by chemist and researchers to handle the access in sucrose production and find other sucrose analogs with new medical and industrial applications (Queneau, Jarosz, Lewandowski, & Fitremann, 2007). Szyszka and co-workers were able to transform sucrose by coupling with a higher amino alditol pendant C-6 of glucose moiety (Szyszka, Osuch-Kwiatkowska, Potopnyk, & Jarosz, 2017). Potewar and et al. were obtained 1,2,3-triazole-sucrose derivatives by applied cycloaddition reaction on sucrose azides and terminal alkynes as antimicrobial, anti fungi derivatives (Potewar, Petrova, & Barros, 2013). Rotsch and co-workers reported the radiosynthesis of 6’-fluoro sucrose (Rotsch et al., 2015). Yi-Xin and co-workers were reported the synthesis of Sucralose-6-acetate via Vilsmeier reaction using SOCl2 in MDF (Yi-dong & Yi-dong , 2013). Zikopoulos and et al. were synthesized different substituent sucrose by halogenated the hydroxyl groups at C-6 and C-6’ of sucrose and investigate their ability to inhabit dextransucrase (Zikopoulos & Nicholas, 1980). Tiara and co-worker synthesized macromolecules by the substitution of terminal positions of sucrose with different unsaturated monosaccharide units (Tiara, Potopnyk, & Jarosz, 2018). Matev and et al. reported the synthesis of sucrose ester by substituted all the primary hydroxyl group with both saturated and unsaturated fatty acid chloride (C10-C10), most of the synthesized sucrose ester showed good antibacterial and antifungal activity (Matev et al., 2013). In contentious with sucrose analogs synthesis, this paper presents new sucrose derivatives by substituted one hydroxyl group with different chain length alkyl halide through by amide linkage via Staudinger reaction. Amide linkage had been chosen for the target compounds because it’s reasonably stable toward pH changing and temperature increased. The hydrophilic head of sucrose core and the hydrophobic tail of the alky group should improve the properties of the target amphiphilic Structures and make them suitable for many bio-applications.
Materials and Methods
Chemicals
Solvents and chemicals were bought form multi companies and were used as they received with no further purifications.
Techniques
TLC was performed on silica gel (Merck GF 254) coated on aluminium plates. The synthesized derivatives compounds were purified by recrystallization using ethanol or by chromatography on silica gel (0.035-0.074 µm) using (hexane: ethyl acetate) or (hexane: acetone) in suitable ratios. The target compounds were dried at 50 ◦C in a vacuum oven over phosphorus pentaoxide prior to physical investigations.
Instruments
1H and 13C NMR spectroscopy were performed in CDCl3, CD3OD and DMSO-d6 on JEOL INM-LA 400. CHN measured by Perkin-Elmer series II 2400-CHNS. Optical rotations were calculated by determining on a Jasco P-1020 digital polar meter using a 10 cm cell. High-resolution mass spectrums were done by LC-Mass Agilent 6530 Q-TOF mass spectrometer.
Synthesis of 6’-O-tert-butyldiphenylsilyl-sucrose [2]
Tert-butylchlorodiphenylsilane (3g,10.91 mmole) was added to sucrose [1] (4 g, 11.69 mmole) in (20 ml) pyridine with a small amount of 4 - Dimethylaminopyridine ( DMAP ) as a catalyst, with stirring at room temperature for about 12h. When TLC indicates the end of the starting material, the solvent was removed to produce (5g, 80%), which is subjected to the next reaction (Barros, Petrova, Correia-Da-Silva, & Potewar, 2011; Karl, Lee, & Khan, 1982).
Acetylating of 6’-O-tert-butyldiphenylsilyl-sucrose [3]
Ethanoic anhydride (15 ml) was added to (5.5 g, 0.000089 mmole) of the derivative [2] in pyridine with continuous stirring at 25 ˚C. 10 hours after, the TLC points to the ending of the reactants. Solvent evaporated. DCM was poured to the mixture then whished by NaHCO3 and water, MgSO4 was used for drying, and the solvent was removed and concentrated to produce (5.5.0g, 72%) of compound [3], which used in the next reaction with no further purification (Lerma & Banik, 2011; Lugemwa, Shaikh, & Hochstedt, 2013; Witschi & Gervay-Hague, 2010).
Desilylation of compound [4]
Tetra-n-butylammonium fluoride (TBAF) (0.06g, 0.025mmole) was added to (5 g, 8.61mmol) tert-butyldiphenylsilyl-sucrose peracetate [3] in (50 ml) THF with stirring at 25 ˚C for 10h. TLC was indicating the consumption of the reactants. THF evaporated, and DCM was added to the crude. The extract was washed with water and dried to yield hepta-O-acetyl-sucrose [4] (3.2 g, 80%) (Gu, Fang, & Du, 2011; Mir & Dudding, 2017; Wang & Demchenko, 2019).
Tosylation of hepta-O-acetyl-sucrose [5]
Tosyl chloride (2ml) was added drop by drop to (6g, 9.42 mmol) of acetylated sucrose [4] in (75 ml) pyridine with stirring for one day. After TLC (hexane-ethyl acetate3:1) shown the consumption of the sugar residue. The mixture added to ice water and separated using DCM. DCM was evaporated to furnished hepta-O-acetyl-6’-O-tosyl-sucrose [5] (5.56 g 75%), which to passed to azidation without further purification (Ananthalakshmi, Kavitha, Kiruthika, & Kalaivani, 2014; Pieper, Schleich, & Gröger, 2019; Yusof & Ariffin, 2016).
Azidation of hepta-O-acetyl-6’-O-tosyl-sucrose [6]
Tosylated sucrose [5] (5.5 g, 6, 96 mmol) and (0.5 g) sodium azide in (75ml) DMF was heated up to about 80-85 ºC. One day later, the TLC showed the finished of the starting material. The mixture was left aside to reach room temperature, extracted with DCM. The organic layer was washed, neutralized and dried with magnesium sulphate. Evaporating the solvent by rotary evaporator to furnish hepta-O-acetyl-6’-O-azido-sucrose [6] (3.54g, 71%) as pure white crystals after recrystallization with ethyl alcohol. (Hosoya et al., 2019; Kamijo, Watanabe, Kamijo, Tao, & Murafuji, 2015; Tomaszewska, Koroniak, & Koroniak, 2017).
Synthesis of hepta-O-acetyl-6’-O-dodecanamido-sucrose [7]
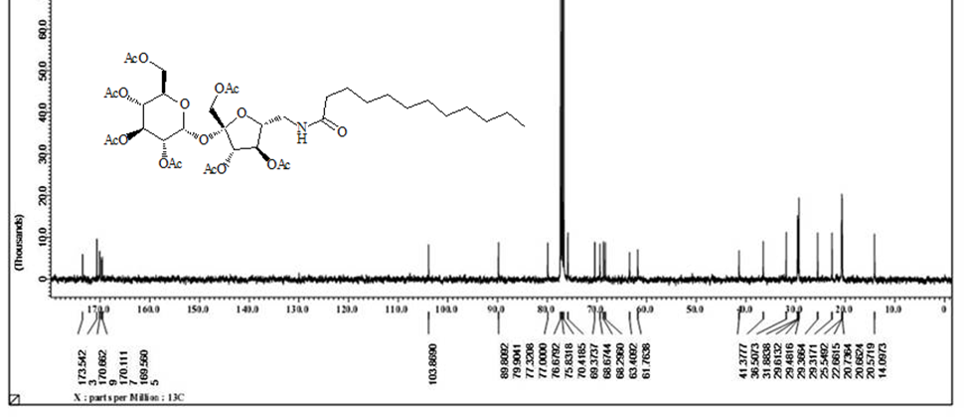
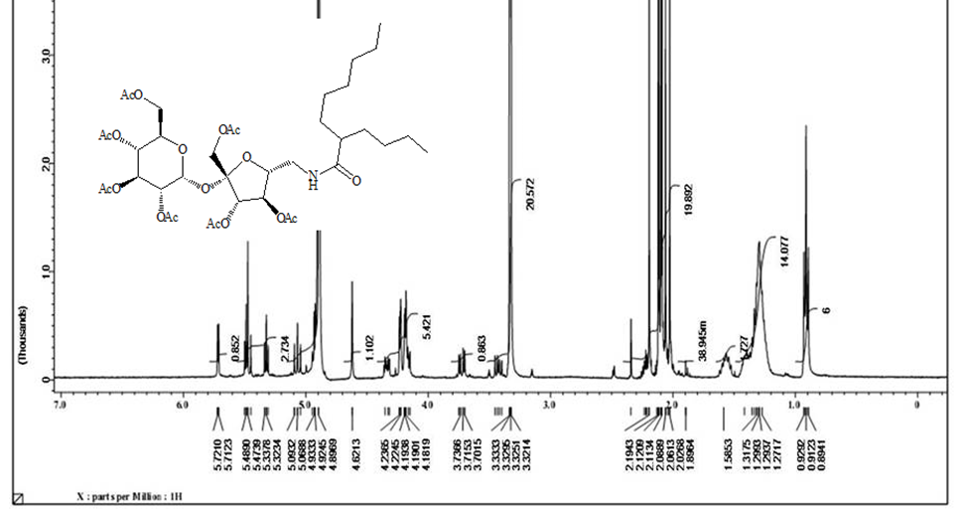
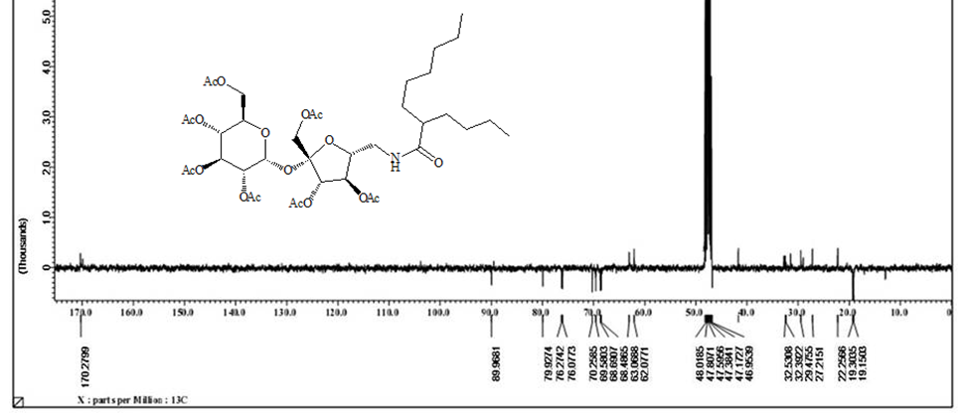
Acetylated sucrose azide [6] (3g, 3.66 mol) reacted with C11H23 COCl and PPh3 by Staudinger coupling to finished (5.2 g, 70%) hepta-O-acetyl-6’-O-dodecanamido-sucrose [7] as NMR pure white crystals. [α]D 25= + 29.0 (C 0.15, chloroform).1H NMR (400 MHz , CDCl3) δ = 5.60 ( d, H-1), 5.40 ( dd, H-3), 5.39 (dd, H-3’), 5.19 (dd, H-4), 4.98 (dd, 4’-H), 4.80 ( dd, H-2), 4.00 ‑ 4.43 ( m, H-1’A , H-1’b, H-5 , H-5’, H-6B, H-6’A), 3.67 (m, H-6A), 3.29 ( m, H-6’B), 2.12 ( m. α-CH2), 2.13 , 2.08, 2.07, 2.06, 2.05, 2.00, 1.99 (7s, 21H, Ac), 1.57 (m, β-CH2), 1.19 ( s, 12H, R group), 0.82 (t, CH3). 3J1,2=3.5, 3J2,3 =10.0, 3J3’,4’=10.0, 2J6 =14.0, 2J6’=13.5, 3J 6’,amide =5.5 Hz Figure 4. 13C NMR (100 MHz, CDCl3) δ =173.53 (C amide), 170,64 x2, 170.10 x2, 195.96, 195.60, 196,55 (C=O), 103.83 (C-2’), 89.78 (C-1), 79.87 ( C-5’), 75.82 ( C-3’), 75.79 ( C-4’), 70.38 ( C-2), 69.33 (C-3), 68.63 (C-5) , 68.29 (C-4), 63.36 (C-6), 61.77 (C-1’), 41.34 (C-6’), 36.47 (α-CH2),31.92 (ω-2), 29.57, 29.63, 29.51, 29.36, 29.28 x2 (R group), 25.53 (β‑CH2), 22.64 (ω-1), 20,74, 20.70, 20.60 x2, 20.59,20.55, 20.553(Ac), 14.00 (ω) Figure 5. CHN for C36H55NO18 calculated: C, 53.32; H, 8.34; N, 2.83; found C 55.37, H 8.40, N 2.87.
Synthesis of hepta-O-acetyl-6’-O-tetradecanamido-sucrose [8]
Acetylated sucrose azide [6] (3.10g, 4.54 mol) reacted with C13H27COCl and PPh3 by Staudinger coupling to produce (6.6g, 69%) of hepta-O-acetyl-6’-O-tetradecanamido-sucrose [8] as NMR pure white crystals. [α]D 25= + 32.0 (C 0.15, chloroform).1H NMR (400 MHz , CDCl3) δ = 5.63 ( d,H-1), 5.43 ( dd, H-3), 5.40 (dd, H-3’), 5.20 (dd, H-4), 5.00 (dd, H-4’), 4.83 ( dd,H-2), 4.04 ‑ 4.45 ( m, H-1’a , H-1’b, H-5 , H-5’, H-6B, H-6’A), 3.69 (m,H-6A), 3.31 ( m,H-6’B), 2.15 ( m. α-CH2), 2.15 , 2.08, 2.09, 2.08, 2.05, 2.02, 2.00 (7s, Ac), 1.59 (m, β-CH2), 1.21 ( s, 16H,R group), 0.84 (t,CH3). 3J1,2=4.0, 3J2,3 =9.5, 3J3’,4’=10, 2J6 =13.5, 2J6’=13.5, 3J 6’,amide =5.0. Hz.13C NMR (100 MHz, CDCl3) δ =173.55 (C amide), 170,66 x2, 170.10 x2, 195.98, 195.63, 196,56 ((C=O), 103.85 (C-2’), 89.79 (C-1), 79.89 ( C-5’), 75.84 ( C-3’), 75.82 ( C-4’), 70.39 ( C-2), 69.35 (C-3), 68.65 (C-5) , 68.30 (C-4), 63.39 (C-6), 61.77 (C-1’), 41.35 (C-6’), 36.49 (α-CH2),31.9 (ω-2), 29.59, 29.60, 29.50, 29.365, 29.29 x2 (R group), 25.54 (β‑CH2), 22.65 (ω-1), 20,75, 20.72, 20.62 x2, 20.63220.59, 20.55(Ac), 14.00 (ω).CHN for C38H59NO18: calculated: C, 55.80; H, 7.27; N, 1.71; found C, 55.85; H, 7.32; N, 1.76.
Staudinger Coupling
Saturated or unsaturated acid halide (1.5 eq.) in of DCM was added as drops to protected sucrose azide [6] (1 eq.) and PPh (1.3 eq) in (25 ml /g) of DCM at 25 ˚C. The cloudy solution was formed after stirred for 12 h at 25 ˚C. The solid separated by filtration, the filtrate washed, neutralized and dried by any available chemicals. The crude resulting after removal of the liquids was purified by on silica gel with 6:1 ethyl acetate- acetone to furnish the acetylated sucrose analogs as pure white crystals (Deketelaere, Nguyen, Stevens, & D’hooghe, 2017; Komjáti, Szokol, Kováts, Kegye, & Nagy, 2018; Ndugire, Wu, & Yan, 2019; Zavar, Zarei, & Saraei, 2016).
Zemplen deacetylating of protected Sucrose
A small amount of CH3ONa was treated with acetylated sucrose amide analogs in a suitable volume of ethanol (30 ml/g). The solution was kept stirring at 25˚C for overnight. When TLC indicated the end of the acetylated sucrose the trace of CH3ONa was destroyed by using Amberlite IR 120 (H+), Filtered and the solvent was removed by an evaporator to produce the final free amide analogs in excellent yield (Fairweather, Liu, Karoli, & Ferro, 2012; Zemplén & Pacsu, 1929; Ágoston, Dobó, Rákó, Kerékgyártó, & Szurmai, 2001).
Synthesis of 6’-O-dodecanamido-sucrose [10]
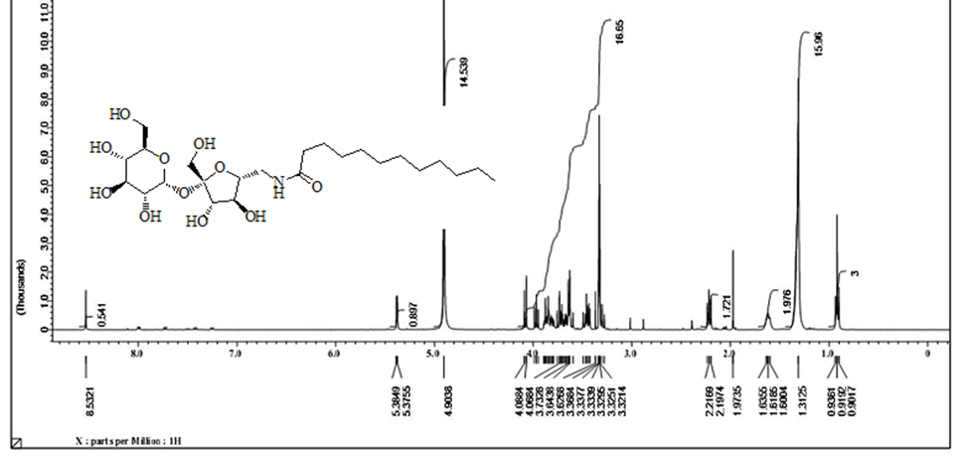
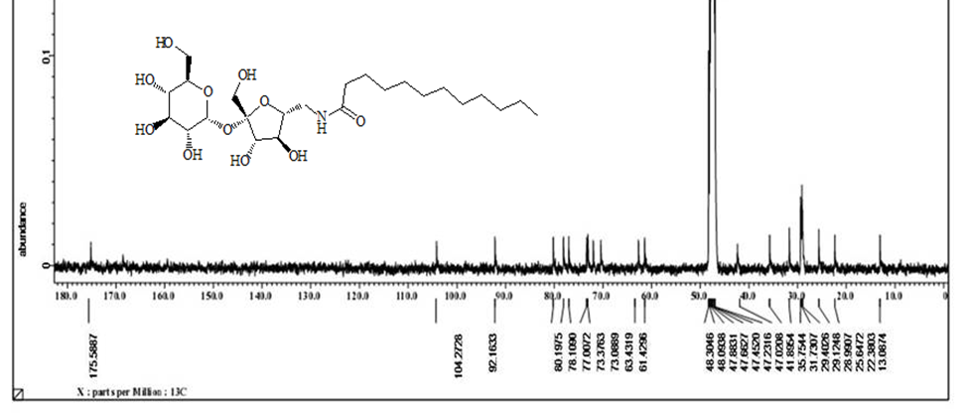
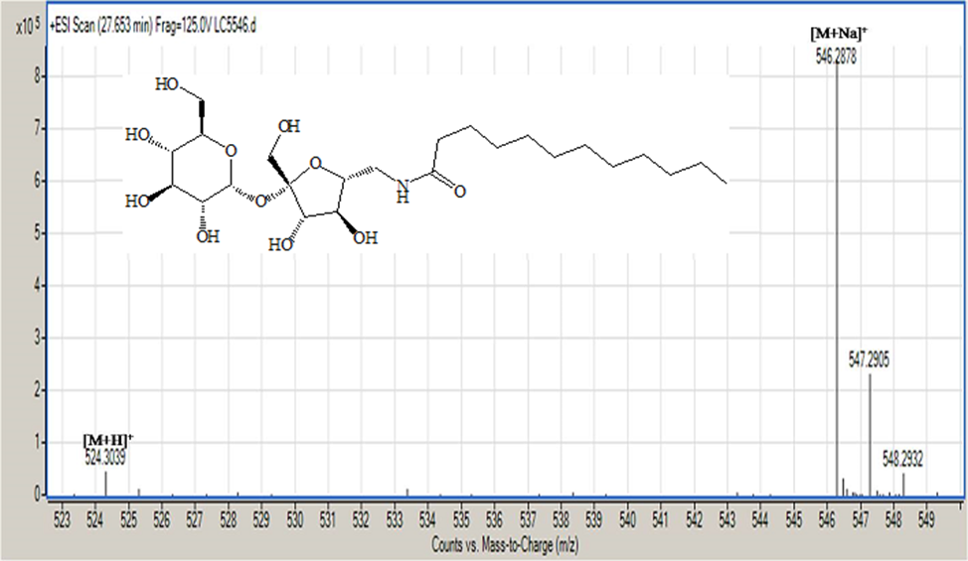
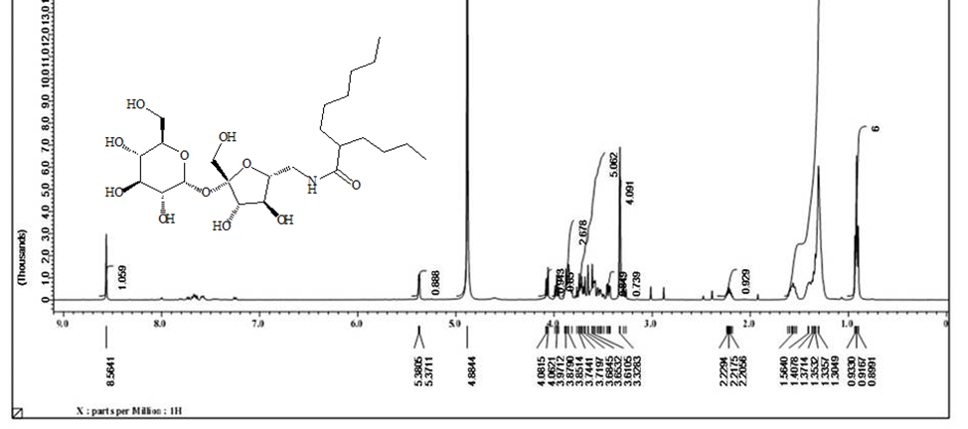
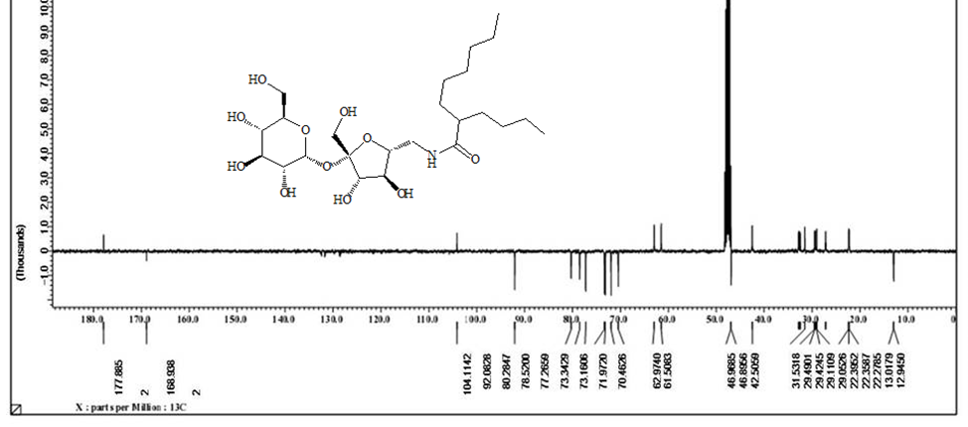
Acetylated sucrose azide [6] (3g, 3.66 mol) reacted with C11H23 COCl and PPh3 by Staudinger coupling to finished (5.2 g, 70%) of hepta-O-acetyl-6’-O-dodecanamido-sucrose [7]. (1 g) of the later was deacetylated using CH3ONa in (30ml) of methyl alcohol as according to Zemplen procedure to yield compound 6’-decanamido-sucrose [10] (0.6 g , 94% ) as white crystals. Melting point (158-160) ͦC.[α]D 25= +37.0 (C 0.15, Ethanol).1H NMR (400 MHz, CD3OD) δ= 3.4 (d, H-1 , 4.05 ( d, H-5’), 3.9- 3.6 (m, H-2’, H-3’ ,H-3 , H-6, H-4, 2H-6’), 2.40 ( dd, H-2), 3.5 (dd, H-1’a), 3.3 (m, H-1’B), 2.19 ( m, α-CH2), 1.59 ( m, β-CH2), 1.29 ( s, 12H, R group), 0.89 ( t, CH3) Hz, 3J1,3 =4, 3J 2,3 =10.5, 2J6=14.00 (ω) Figure 6. 1H NMR (400 MHz, CD3OD) δ= 175.6 (CO amide), 104.65 ( C-2’), 92.15 (C-1), 80.20 (C-3’), 78.05 (C-5’ , 76.99 (C-5), 73.35 (C-3) , 73.06 (C-4’), 72.07 (C-2), 70.63 (C4), 63.22 (C-6), 61.40 (C-1’), 42.30 (C-6’) ,35.76 (α-CH2), 31.72 (ω -2), 29.39 x3, 29.10 x2, 29.0 x3, 28.92 x2 ( R group), 25.62 (β-CH2), 22.37 (ω-1), 13.06 (ω) Figure 7. CHN for C22H41NO11: calculated C, 53.32; H, 8.34; N, 2.83: found; C, 53.38; H, 8.40; N, 2.87. HRMS: [M+H]+: calcd for C24H45NO11: 524.3065 (100%) , 525.3098 (28 %); found : 524.3039 ( 100 %), 525.3064 ( 25 %) ;[M+Na]+: calcd for C24H45NO11Na: 546.2884 (100%), 547.2917 (28% ; found : 546. 2878 (100%), 547.2905 (28%). (For CHN and NMR and HRMS analysis and spectrumsof the acetylated analog [7], Figure 9.
Synthesis of 6’-O-tetradecanamido-sucrose [11]
Acetylated sucrose azide [6] (3.10g, 4.54 mol) reacted with C13H27COCl and PPh3 by Staudinger coupling to produce (6.6g, 69%) of hepta-O-acetyl-6’-O-tetradecanamido-sucrose [8]. (1 g) of the later was reacted with CH3ONa in (30ml) of methyl alcohol via Zemplen procedure to yield 6’-O-tetradecanamido-sucrose [11] (0.6 g , 94% ) as white crystals. Melting point (184-186) ˚C .[α]D 25= +41.0 (C 0.15, Ethanol).1H NMR (400 MHz, CD3OD) δ= 3.4 (d, H-1 , 4.06 ( d, H-5’), 3.9- 3.58 (m, H-2’, H-3’ ,H-3 , H-6, H-4, 2H-6’), 2.41 ( dd, H-2), 3.47 (dd, H-1’A), 3.27 (m, H-1’B), 2.20 ( m, α-CH2), 1.60 ( m, β-CH2), 1.30 ( s, 16H, R group), 0.90 ( t, CH3) Hz, 3J1,3 =3.5, 3J 2,3 =10.5, 2J6=13.0) Figure 6.1H NMR (400 MHz, CD3OD) δ= 175.57 (C amide), 104.66 ( C-2’), 92.15 (C-1), 80.20 (C-3’), 78.9 (C-5’ , 77.01 (C-5), 73.38 (C-3) , 73.09 (C-4’), 72.07 (C-2), 70.65 (C4), 63.22 (C-6), 61.41 (C-1’), 42.28 (C-6’) ,35.76 (α-CH2), 31.72 (ω -2), 29.41 x3, 29.11 x2, 28.98 x3, 28.93 x2 ( R group), 25.63 (β-CH2), 22.39 (ω-1), 13.06 (ω).Figure 7 CHN C24H45NO11: Calculated. C, 55.05; H, 8.66; N, 2.68; found: C, 55.11; H, 8.71; N, 2.73. (For CHN and NMR analysis of the acetylated analog [8] sees the supplementary materials)
Synthesis of hepta-O-acetyl-6’-O-(2-butyl-octanamido)-sucrose [12]
Acetylated sucrose azide [6] (2.g, 4.5 mol) treated with (Octanoyl chloride, 2-butyl- ) and PPh3 by Staudinger coupling to give(2.g, 65%) of hepta-O-acetyl-6’-O-(2-butyl-octanamido)-sucrose [12], as NMR pure white crystals.[α]D 25= + 25.0 (C 0.15, chloroform).1H NMR (400 MHz , CD3OD) δ = 5.18( d, H-1), 5.7 ( dd, H-3), 5.50 (dd, H-3’), 4.98 (dd, H-4 , 5.29 (dd, H-4’), 4.92 ( dd, H-2), 4.16-4.20 ( m, H-1’B, H-5 , H-5’ , H-6B, H-6’A), 3.69 (m, H-6A) ,2.19 ( m, α-CH2), 3.29 (-CH), 2.19, 2.09, 2.108 ,2.07, 2.06, 2.05. 1.99 (7s, Ac), 1.6 (m, β-CH2), 1.29 ( m, 16H,R group), 0.89 (t, CH3). 3J1,2=4.0, 3J2,3 =9.5, 3J3’,4’=10.0, 2J6 =1.5, Hz Figure 10.13C NMR (100 MHz, CD 3 OD) δ = 177.3 (C amide), 170,.39, 170.35 x2, 170.29, 197.00, 196,86 (C=O), 88.91 (C-2’), 79.90 (C-1), 76.25( C‑5’), 75.98 ( C-3’), 70.21 ( C-4’), 69.5 ( C-2), 68.5 (C-3), 68.3 (C-5) , 68.00 (C-4), 63.00 (C-6), 62.05 (C-1’), 41.69 (C-6), 32.86. 32.73 (α-CH2),29.58, 29.56, 32.50, 32.35, 31,40 (R group), 31.39, 31.29 (β-CH2), 22.63 (ω-1), 29.50, 29.00, 28.95 (Ac), 19.30, 19.13 (ω)Figure 11. CHN for C38H59 NO18: calculated C55.81, H 7.27, N 1.71; found C 55.85, H 7.38, N 1.74.
Synthesis of hepta-O-acetyl- 6’-O-(2-hexayl-decanoyl)-sucrose [13]
Acetylated sucrose azide [6] (3.20.g, 4.5 mol) treated with (decanoyl chloride, 2-hexyl- ) and PPh3 by Staudinger coupling to yield (4g, 65%) of hepta-O-acetyl- 6’-O-(2-hexayl-decanoyl)-sucrose [13] as NMR pure white crystals.[α]D 25= + 27.0 (C 0.15, chloroform).1H NMR (400 MHz , CD3OD) δ = 5.13 ( d, H-1), 5.48 ( dd, H-3), 5.48 (dd, H-3’), 4.97 (dd, H-4 , 5.28 (dd, 4’-H), 4.89 ( dd, 2‑H), 4.16-4.34 ( m, H-1’B, H-5 , H-5’ , H-6B, H-6’A), 3.68 (m, H-6A) ,2.17 ( m, α-CH2), 3.27 (-CH), 2.17, 2.13, 2.10 ,2.08, 2.07, 2.06. 2.00 (7s, Ac), 1.4 (m, β-CH2), 1.27 ( m, 16H,R group), 0.92 (t, CH3). 3J1,2=4.0, 3J2,3 =9.5, 3J3’,4’=10.0, 2J6 =1.5, Hz.13C NMR (100 MHz, CD 3 OD) δ = 177.4 (C amide), 170,.39, 170.34 x2, 170.31, 196.88, 196,92 (C=O), 88.95 (C-2’), 79.88 (C-1), 76.23( C‑5’), 76.03 ( C-3’), 70.21 ( C-4’), 69.58 ( C-2), 68.5 (C-3), 68.4 (C-5) , 68.2 (C-4), 62.98 (C-6), 62.06.CHN for C42H67NO18 : calculated ; C, 57.72; H, 7.73; N, 1.60: found; C, 57.78; H, 7.79; N, 1.66.
Synthesis of 6’-(2-Butyl-octanamido)-sucrose [14]
Acetylated sucrose azide [6] (2.g, 4.5 mol) treated with (Octanoyl chloride, 2-butyl- ) and PPh3 by Staudinger coupling to give (2.g, 65%) hepta-O-acetyl-6’-O-(2-butyl-octanamido)-sucrose [12]. (2 g) of the later was reacted with CH3ONa in (30ml) of methyl alcohol via Zemplen procedure to provide (1.18 g, 92%) of 6’-(2-Butyl-octanamido)-sucrose [14] as white crystals. Melting point (149-151) ˚C [α]D 25= + 43.0 (C 0.15,Ethanol).1H NMR (400 MHz, CD3OD) δ= 5.40 (d, H-1), 4.39 (d, H-5’), 3.90- 3.50 (m, H-2’, H-3’, H-3, H-6, H-4, 2H-6’), 3.45(m, H-2), 3.30 (m, H-1’a, 1’b), 2.19 (m, α-CH2), 1.57 ( m, β-CH2), 1.33 ( s, 16H, R group), 0.89 ( t, CH3) Hz, 3J1,3 =4.0, 3J 2,3 =9.5 , 2J6=14.0) Figure 12.1H NMR (400 MHz, CD3OD) δ= 177.90 (C amide), 104.10 (C-2’), 92.07(C-1), 80.30 (C-3’), 78.50 (C-5’), 77.25 (C-5), 73.35 (C-3), 73.15 (C-4’), 72.0(C-2), 70.45 (C4), 63.00 (C-6), 61.48 (C-1’), 42.49 (C-6’),32.85, 32.75, 32.52, 32.45 (α-CH2), 31.52 x2 (ω-2), 29.50 x3, 29.40, 29.10, 29.0 (bulk-CH2), 27.20, 27.15 (β-CH2), 22.40, 22.36, 22.70(ω-1), 13.00, 12.95 (ω) Figure 13. CHN for: C24H45NO11 ; calculated. C, 55.05; H, 8.66; N, 2.68: found: C, 55.09; H, 8.74; N, 2.72. HRMS: [M+H]+: calcd for C24H45NO11: 524.3007 (100%) , 525.3000 (28 %); found : 524.3159 (very weak) ;[M+Na]+: calcd for C24H45NO11Na: 546.2880 (100%) , 547.2920 (28%); found : 546. 2990 (100%), 547.3020 (24%). (For CHN and NMR analysis of the acetylated analog [12]
Synthesis of 6’-(2-Butyl-octanamido)-sucrose [15]
Acetylated sucrose azide [6] (3.20.g, 4.5 mol) treated with (decanoyl chloride, 2-hexyl- ) and PPh3 by Staudinger coupling to yield (4g, 65%) of hepta-O-acetyl- 6’-O-(2-hexayl-decanoyl)-sucrose [13]. (2.4 g) of the later was reacted with CH3ONa in (30ml) of methyl alcohol via Zemplen deacetylating procedure to yield 6’-(2-Butyl-octanamido)-sucrose [15] (1.25 g, 92%) as white crystals. Melting point (175-177) ˚C [α]D 25= + 43.0 (C 0.15,Ethanol).1H NMR (400 MHz, CD3OD) δ= 5.40 (d, H-1), 4.39 (d, H-5’), 3.90- 3.50 (m, H-2’, H-3’, H-3, H-6, H-4, 2H-6’), 3.45(m, H-2), 3.30 (m, H-1’a, 1’b), 2.19 (m, α-CH2), 1.57 ( m, β-CH2), 1.33 ( s, 16H, R group), 0.89 ( t, CH3) Hz, 3J1,3 =4.0, 3J 2,3 =9.5 , 2J6=14.0).1H NMR (400 MHz, CD3OD) δ= 177.90 (C amide), 104.10 (C-2’), 92.07(C-1), 80.30 (C-3’), 78.50 (C-5’), 77.25 (C-5), 73.35 (C-3), 73.15 (C-4’), 72.0(C-2), 70.45 (C4), 63.00 (C-6), 61.48 (C-1’), 42.49 (C-6’),32.85, 32.75, 32.52, 32.45 (α-CH2), 31.52 x2 (ω-2), 29.50 x3, 29.40, 29.10, 29.0 (bulk-CH2), 27.20, 27.15 (β-CH2), 22.40, 22.36, 22.70(ω-1), 13.00, 12.95 (ω).CHN for: C24H45NO11 ; calculated. C, 55.05; H, 8.66; N, 2.68: found: C, 55.09; H, 8.74; N, 2.72. (For CHN and NMR analysis of the acetylated analog [13]
Results and Discussion
Synthesis
The resent work is aim to synthesis new biodegradable amide derivatives from sucrose by modified one of its (8) hydroxyl groups, as the hydroxyl groups at (C-6, C-6’, C-1’) are primary hydroxyl groups. There is a good opportunity to modify one those hydroxyl groups for the easy synthesis of the great and huge derivatives that have multi applications. C-6' hydroxyl groups were chosen due to the availability of sources and chemical reagents. Two couples of fatty acids (C12, C14, C8-4, C10-6) were selected for the coupling with sucrose at C-6’ position by amide linkage via Staudinger reaction.
The synthetic scheme for this synthesis illustrated step by steps in (Figure 1) shown below. The eight alcoholic groups in the structure of sucrose make a single coupling with only one alcoholic group is difficult to achieve, but still available by depending on the difference in reactivity of the hydroxyl group. Based on this reactivity, the primary hydroxyl group is more active then secondary ones, and the primary hydroxyl group at carbon number of glucose moiety C6-OH is the same activity of the primary hydroxyl group at carbon six-position of fructose C'6-OH, while the primary hydroxyl group at carbon C1-OH of fructose coming last.
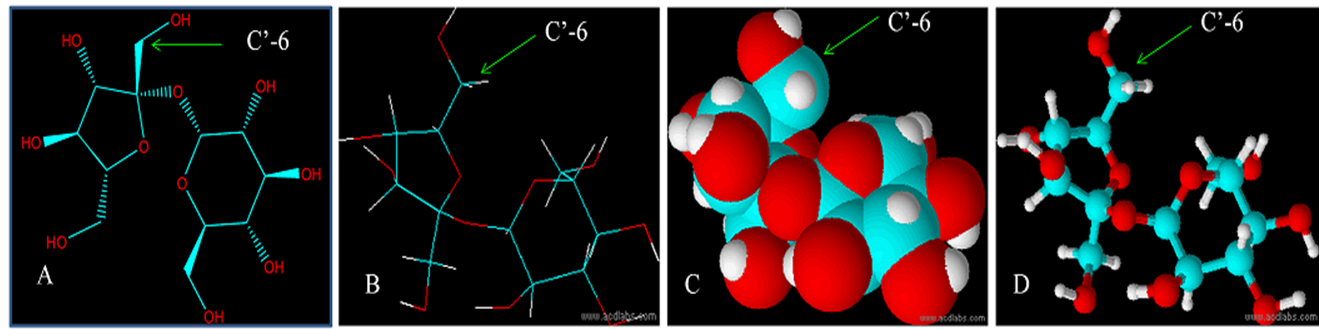
The competition is still between hydroxyl groups at C6-OH and C’6-OH, The 3D structure of sucrose (Figure 2) shown the steric hindrance at C’6-OH is less then C6-OH, so that the priority of reaction can be handled by directed the reaction toward C'6-OH by using bulky reagents.
The use bulky reagent butyldiphenylchlorosilane can break the competition and controlling the reaction by directed it toward C’6-OH, which is led to the unique protection of the alcoholic group at position six-carbon of the fructose part and produced [2]. Treatment of the later with acetic anhydride in pyridine introduce full protected analog [3].
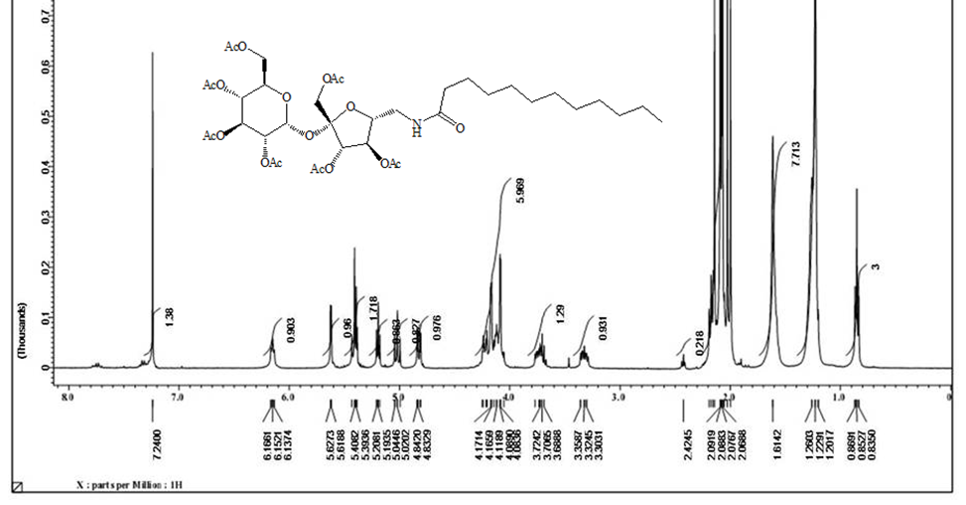
The solo protected group at C’6-OH can be free again using tetra-n-butylammonium fluoride to produced compound [4] with an only free hydroxyl group. The unique free hydroxyl group was converted to a good leaving group by the reaction with tosyl chloride in pyridine to give compound [5] . The later [5] is subjected to the reaction with sodium azide in pyridine to get the precursor of the target synthesis [6] . A Series from four acetylating compounds have been synthesized from compound [6] by the reaction with four fatty acids chloride (C12, C14, C8-4, C10-6) using Staudinger coupling to produce [7,8,12,13] in protected form. Treatment of those compounds with sodium methoxide in methanol via Zemplen reaction free all the alcoholic groups and yields the final derivative [10,11,14,15]. Elemental analysis CHN for the final compounds approximately fitted the theoretical calculation except for little deviation in hydrogen value, which indicates the hygroscopic properties of the final products. Both 1H NMR and 13C NMR have proven the purity and the chemical structures of the final compounds. 1H NMR analysis are done based on the acetylated compound as the protection prevent the peaks overlapping which appears clearly in deacetylating compounds. Acetylating derivatives show the signal of the amide linkage at around δ( 6.6), the protons of sucrose appears in between δ (3.6-5.8), acetyl groups signals are located at around δ 1.98, and the alkyl groups signals are listed in between δ (2.5- 0.84). While 13C NMR signals for amide linkage from δ (171-17)5, fructose moiety of sucrose between δ (195-198) glucose part of sucrose δ (196.5-197), sugar carbon δ (62) for C-1’- δ (104) for C-2’, C-6’ at δ (42) and R group appear in between δ (37) for α-CH2 and δ (14.5) for CH3. The free derivatives signals appear around δ (7.75 ) for amide protons, sugar moiety protons in between δ (3.98-3.5) and the protons of the R groups located in between δ (2.20) for α-CH2 and δ (0.91) for CH3. The 13C NMR show the amide linkage around δ (175.4) and sugar signals from δ (61.2) for C-1’ to δ (92) for C-1. The primary carbons of furanose located around δ (42) for C-6’ and the R group from δ (36) for α-CH2 to δ (13-35) for CH3.
Physiochemical Properties
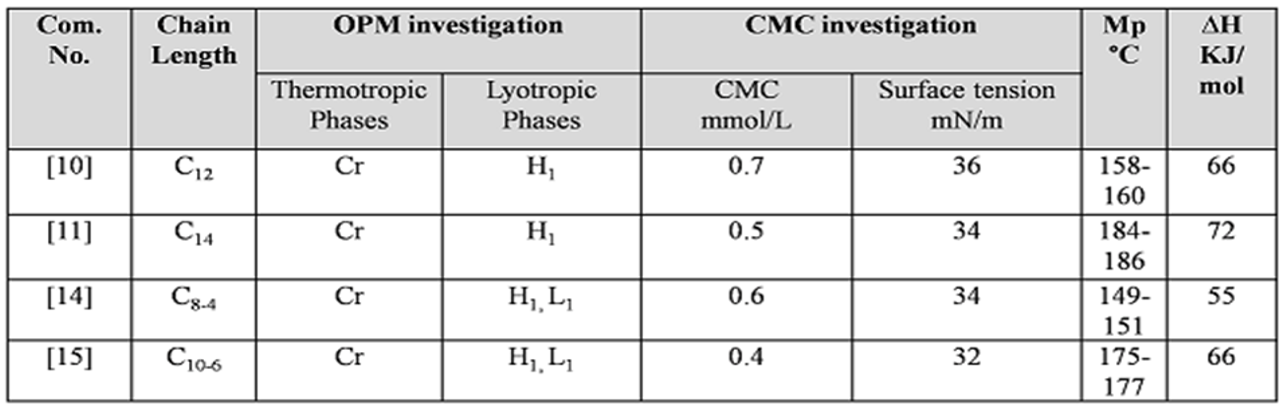
Physiochemical properties are summarized in Figure 3. All of the sucrose derivatives are soluble in water at room temperature. Sucrose compounds are exhibit high melting point, which translates the high interaction between the hydrophilic head group of sucrose moiety and the hydrophobic interaction between the long-chain alkyl group.
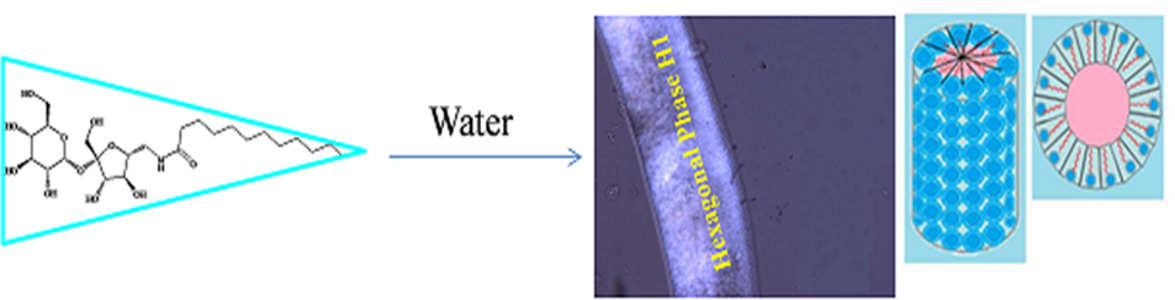
The target derivatives show only crystalline in thermotropic phase, while the hexagonal phase clearly appears in all compounds in penetrating with water as a lyotropic phase and this phase is expected due the as the head group of sucrose moiety is too large in comparing with small hydrophobic alkyl chain Figure 8.
The surface tension was measured for the series of sucrose compounds solution with different concentrations by the DuNouy ring The critical micelles concentrations CMC of the sucrose monoamide in aqueous solutions were evaluated from the across a point in surface tension versus log concentration plots at 25°C (Nouy, 1919). All sucrose derivatives are shown an ability to reduce the surface tension with different degrees, and the results are in a good agreement with other sugar surfactants (Petkova, 2018; Zhang, Song, Taxipalati, Wei, & Feng, 2014). The ability to reduce the surface tension increased with the increasing number of the carbon atoms in the hydrophobic group 32 mN/m for C10-6 to 36 mN/m for C12 , the ability of reducing surface tension, the solubility at room temperature and biodegradability make the sucrose compounds a good candidate for oil in water emulsifier.
Conclusion
Sucrose monoamide derivative can be achieved in good yield based on the comparison of the activity of the hydroxyl groups and control the priority for the target position of reaction via using protection groups. Followed by activation of the target hydroxyl group, functionlization then coupling with the corresponding halide alkyl. The final amide produced after removing the protection group. Final characterization and proved the purity and structure by available means for the final products which is considered can a good candidate as surface-active agents.