In Silico studies of 4-Anilino Quinazoline derivatives as Anti-tubercular Agents
Abstract
Inh A, the Enoyl Acyl Carrier protein Reductase from Mycobacterium tuberculosis is one of the pivotal enzyme involved in the mycobacterial fatty acid elongation cycle and has been considered as an important target for anti-tubercular screening. Inhibition of Inh A affects the biosynthesis of the mycolic acids, which are the central constituents of the mycobacterial cell wall. In the present research work, 4-anilino quinazoline derivatives were designed based on the quinazoline based drugs by means of lipophilic insertion and Fragment replacement. The designed compounds were synthesized, and molecular docking studies were performed on the human pathogenic bacterial enzyme InhA from its parent domain Mycobacterium Tuberculosis. Molecular docking study revealed that compounds SMOQ2, SNAQ3, 4AAQ7, 2AP9, PABAQ10 were found to possess good binding affinity towards the target InhA. With reference to the binding energy obtained from molecular docking study, five compounds were subjected to in vitro anti-tubercular activity against M. tuberculosis H37Rv and I2487 (Resistant strain) using BACTEC MGIT method. Compound SMOQ2 and 4AAQ7 showed sensitivity in both H37Rv (Sensitive strain) and I2487 (Resistant strain) at the concentration of 250, 500, 1000 and 1500 mcg/mL. In silico Pharmacokinetic predictions of the synthesized compounds were determined using SwissADME online web tool. All the synthesized compounds obeyed the Lipinski's rule of five properties.
Keywords
Enoyl-acyl Carrier Protein Reductase, 4-anilino quinazoline, Mycobacterium Tuberculosis, BACTEC MGIT method
Introduction
Mycobacterium tuberculosis, the causative agent of tuberculosis (TB), responsible for the morbidity and mortality of a large population worldwide (Franco, Martínez, Rodríguez, & Wertheimer, 2009). In 2015, WHO estimated that there were 580,000 new cases of MDR-TB and that 250,000 MDR-TB deaths occurred globally. About 10% of healthy individuals may develop TB in their lifetime due to various genetic factors (Barrett & Barrett, 2003). The development of drug resistance against various anti-tubercular drugs and the influence of HIV epidemic has made the disease remain a major global public health problem (Jiang et al., 1990). According to WHO, one-third of the world's population have been infected with Mycobacterium tuberculosis (MTB) (Xia et al., 2001). Inh A, the Enoyl Acyl Carrier protein Reductase from Mycobacterium tuberculosis is one of the enzymes which is involved in the mycobacterial fatty acid elongation cycle. Inh A is considered as the main target for anti-tubercular drug designing. Inhibition of Inh A affects the biosynthesis of the mycolic acids, which is central constituents of the mycobacterial cell wall. Hence the cell wall synthesis gets interrupted.
Quinazolines are classes of fused heterocyclic ring system with a broad spectrum of pharmacological activities such as anti-cancer, anti-tubercular, anti-bacterial (Gangwal, Kothawade, Galande, Pharande, & Dhake, 2001), anti-fungal (Bartroli et al., 1998), anti-HIV (Alagarsamy et al., 2004), anthelmintic (Gupta, Kumar, & Ahmad, 1987), analgesic (Manivel, Naveenraj, Kumar, & Anandan, 2010), anti-inflammatory (Chao et al., 1999), anti-hypertensive (Wright, Tomcufcik, Chan, Marsico, & Press, 1987), anti-diabetic (Saeedi et al., 2019) and anti-oxidant (Subramaniam, Pai, Vigra, & Sodhi, 2010) activities.
The present work involves the molecular interactions of the Enoyl-Acyl carrier protein Reductase Mycobacterium tuberculosis (InhA), (PDB ID: 4TZK) with the designed ligands using molecular docking tool. With reference to the binding energy, compound SMOQ2, SNAQ3, 4AAQ7, 2APQ9 and PABAQ10 were evaluated for their in vitro anti-mycobacterial activity against Mycobacterium tuberculosis H37Rv (Sensitive stain) using BACTEC MGIT method. Compound SMOQ2 and 4AAQ7 showed sensitivity in both H37Rv (Sensitive stain) and I2487 (Resistant strain) at the concentration of 250, 500, 1000 and 1500 mcg/mL. The designed derivatives were predicted for their in silico ADME predictions using the SwissADME server.
Materials and Methods
Chemistry
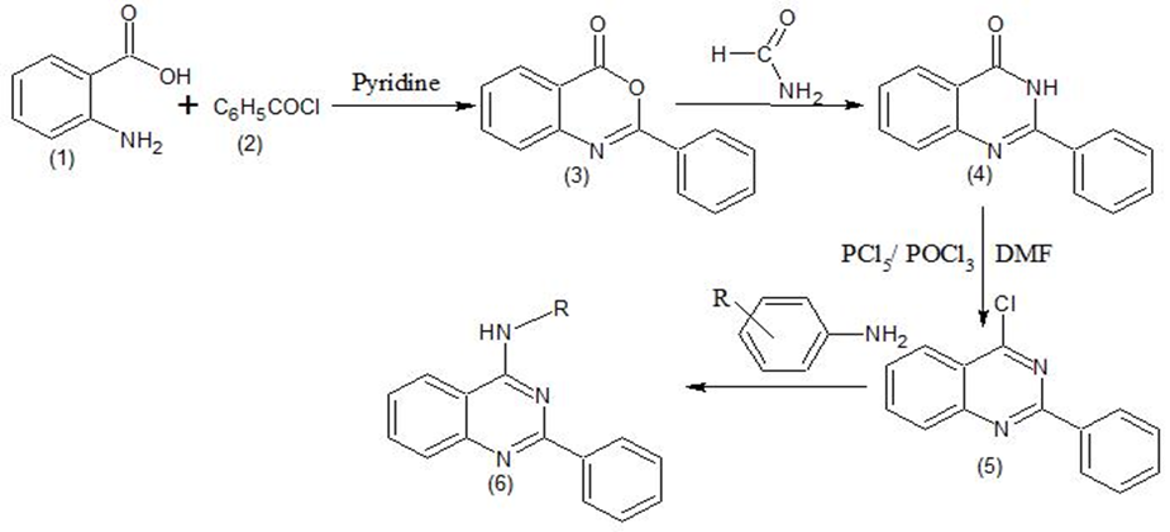
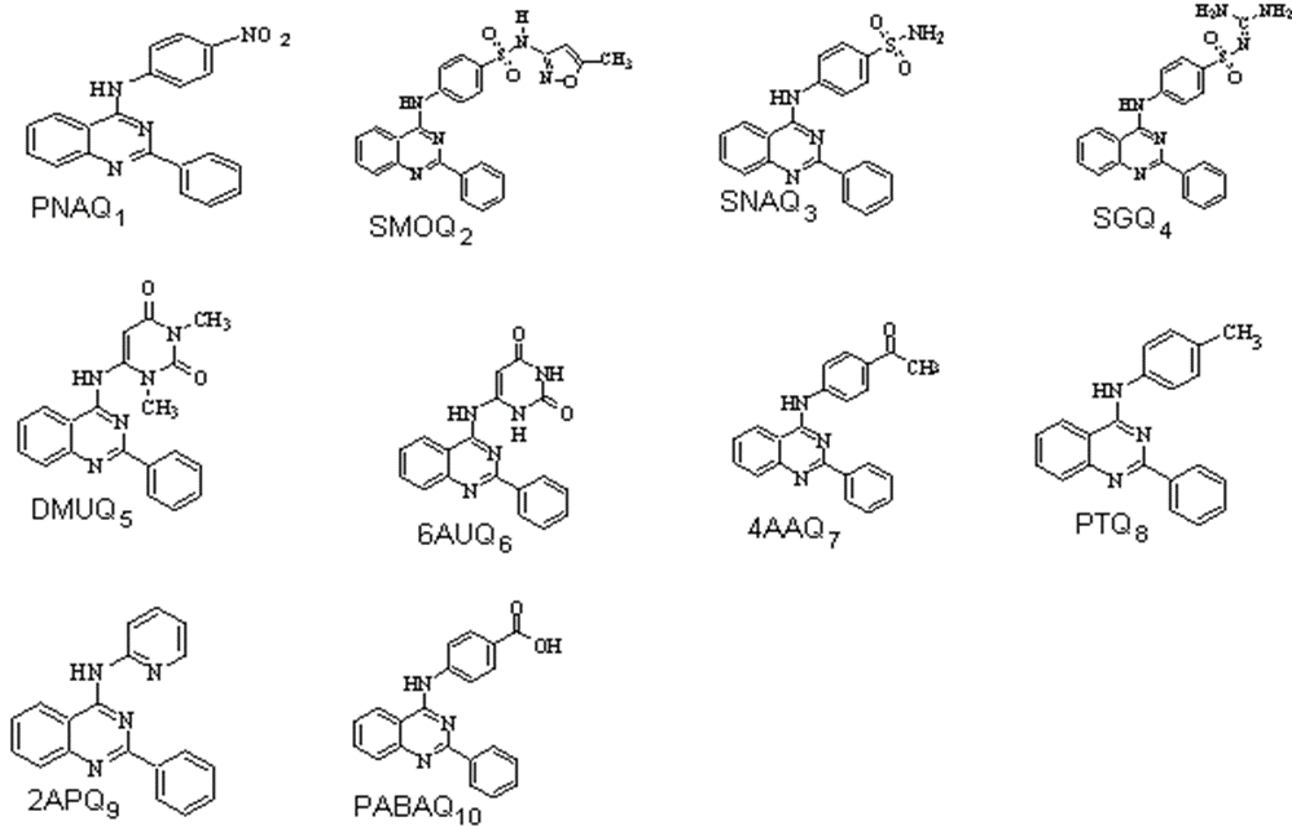
A series of 4-Anilino quinazoline derivatives have been designed (Figure 1) and synthesized from anthranilic acid in four steps via benzoxazinones, Quinazolin-4-ones and 4-Chloro quinazolines. The synthetic work has been published. (Hemalatha, Selvin, & Girija, 2018) The scheme for the synthesis of title compounds were depicted in Figure 1 and their structure were given in Figure 2.
Molecular Docking
Protein Preparation
Molecular docking was performed for 4-Anilino quinazolines using AUTODOCK 4.0 on the Windows 64-bits operation system. The target protein Enoyl-Acyl carrier protein Reductase Mycobacterium tuberculosis (InhA), (PDB ID: 4TZK) in complex with 1-cyclohexyl-N-(3,5-dichlorophenyl0-5-oxopyrrolidine-3-carboxamide were retrieved from Protein Data Bank (He, Alian, Stroud, & Montellano, 2006).
Ligand Preparation
Chemsketch was used to draw the structures of designed Quinazoline derivatives which acts as a ligands followed by generation of 3D structure in PDB format using Marvin sketch.
The structure with the lowest binding free energy was chosen for the optimum docking conformation and interaction with the target.
Absorption, Distribution, Metabolism and Excretion (ADME) prediction
ADME screening were checked to predict the various physiochemical and pharmacokinetic properties of the designed 4-anilino quinazoline derivatives and also to predict their drug-likeness properties. In silico pharmacokinetic screening was done using the online SwissADME web tool. MW- Molecular Weight, RB- No. of Rotatable bonds, TPSA- Topographic Polar Surface Area, ESOL- Water solubility, SOL-Soluble, M.SOL- Moderately Soluble, V. SOL- Very Soluble, GIA- GastroIntestinal Absorption, BBB P- Blood-Brain Barrier Permeability, PgpS- Permeability Glycoprotein Substate, CYP1- CYP9 inhibitor, CYP2, a CYP2D6 inhibitor, LogKp (cm/s)-Skin permeant
Evaluation of Anti-tubercular Activity
Based on the binding energy obtained from the molecular docking study (least energy), 5 compounds SMOQ2, SNAQ3, 4AAQ7, 2APQ9 and PABAQ10 were subjected to in vitro anti-tubercular study by MGIT (mycobacteria growth indicator tube) sensitivity method at various concentrations 250, 500, 1000, 1500 mcg/mL against the sensitive strain (H37Rv) and Resistant Strain (I2487). Positive tubes, identified by the BD BACTED MGIT instrument, should be sub-cultured and an acid-fast smear prepared.
Results and Discussion
Molecular Docking Study
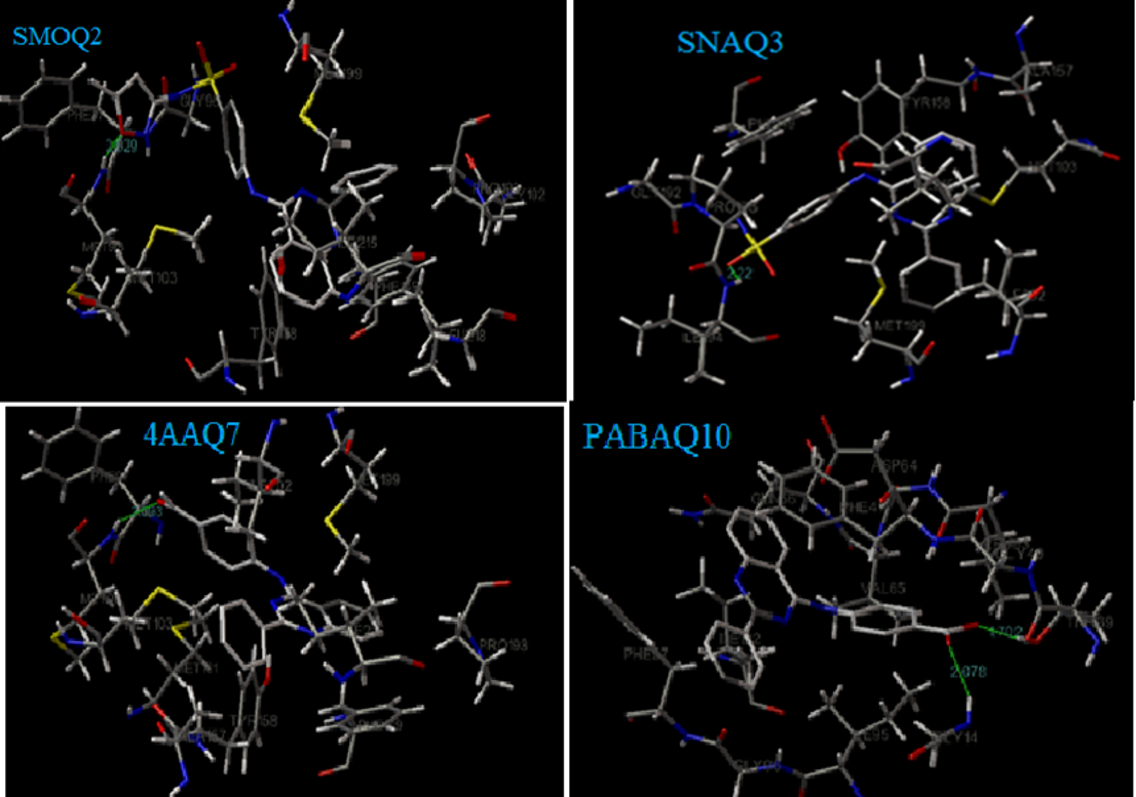
Molecular docking study showed that compound SMOQ2 (-9.66 kcal/mol), SNAQ3 (-9.31 Kcal/mol), 2APQ9 (-9.19 kcal/mol), 4AAQ7 (-8.85 Kcal/mol) and PABAQ10 (-8.8 kcal/mol) possess the highest binding affinity towards the target InhA compared to the standard drug Isoniazid (-5.6 Kcal/mol). The compound with least negative binding free energy means a better binding; The free energy of interactions/binding for each compound were compared to standard Isoniazid. Based on these results, Isoniazid with the binding free energy of -5.6kcal/mol can form two hydrogen bonds with amino acid residues Val 65 and Gly 14 with the distance of 2.014 A° and 2.061 A° respectively. Besides, Isoniazid showed van der Waals interactions with Ile 95, Thr 39, Leu 63 and Phe 41. Compound SMOQ2 showed the binding energy of -9.66 Kcal/mol and formed one hydrogen bond with amino acid residue Met 98 (NH) with the distance of 2.029 A°, followed by Compound SNAQ3 showed the binding energy of -9.31 Kcal/mol and formed one hydrogen bond with the amino acid residue Ile 194 (NH) and its interatomic distance was 2.22 A°. The docking results were shown in Table 1. The inhibition constant, vander waals desolvation energy and the various amino acid involved in vander waals interactions were given inTable 2. The molecular interactions of designed ligands in the active site of the protein were shown in Figure 3.
|
Sl.No. |
Compound code |
No. of hydrogen bond formed |
Amino acid involved in Hydrogen bond interactions |
Distance between Donor and Acceptor (Å) |
Binding Energy |
|---|---|---|---|---|---|
|
1 |
PNAQ1 |
1 |
Thr 158 |
2.173 |
-8.51 |
|
2 |
SMOQ2 |
1 |
Met 98 (NH) |
2.029 |
-9.66 |
|
3 |
SNAQ3 |
1 |
Ile 194 (NH) |
2.22 |
-9.31 |
|
4 |
SGQ4 |
1 |
Pro 156 |
2.868 |
-8.64 |
|
5 |
DMUQ5 |
1 |
Gly 14(NH) |
2.119 |
-6.13 |
|
6 |
6AUQ6 |
2 |
Ile 15 Gly 14(NH) |
2.21 2.075 |
-8.64 |
|
7 |
4AAQ7 |
1 |
Met 98 (NH) |
2.093 |
-8.85 |
|
8 |
PTQ8 |
1 |
Met 98 (NH) |
2.088 |
-8.16 |
|
9 |
2APQ9 |
2 |
Pro 156 (O) Pro 156 (O) |
2.694 2.994 |
-9.19 |
|
10 |
PABAQ10 |
2 |
Gly 14 Thr 39 |
2.078 1.702 |
-8.8 |
|
11 |
Standard (Isoniazid) |
2 |
Val 65 (NH) Gly 14 (NH) |
2.014 2.061 |
-5.6 |
|
Sl. No. |
Compound code |
Inhibitory constant (μM) |
Vdw. Desolvation Energy |
Aminoacid involved in van der waals interactions |
|---|---|---|---|---|
|
1 |
PNAQ1 |
577.61 |
-9.35 |
Leu 218, Phe 149, Pro 193, Tyr 158, Ile 215, Met 161, Ala 167, Gly 104, Met 103, Leu 207, Met 98, Phe 97, Ile 202, Met 199 |
|
2 |
SMOQ2 |
125.61 |
-11.2 |
Leu 207, Ile 202, Met 199, Met 103, Met 161, Ala 157, Tyr 158, Ile 216, Pro193, Phe 149, Asp 148, Met 155, Leu 216, Ala 191 |
|
3 |
SNAQ3 |
148.77 |
-10.52 |
Ile 194, Gly 192, Pro 193, Phe 149, Met 199, Tyr 158, Ile 215, Ile 202, Ala 157, Met 103 |
|
4 |
SGQ4 |
632.78 |
-9.17 |
Ile 95, Val65, Thr 39, Phe 97, Gly 96, Gly 40, Ile 47, Phe 41 |
|
5 |
DMUQ5 |
348.22 |
-6.55 |
Thr 39, Ser 94, Gly 96, Ile 95, Val 65, Phe 97, Leu63 |
|
6 |
6AUQ6 |
461.45 |
-9.53 |
Ile95, Val 65, Thr 39, Phe 97, Gly 96, Gly 40, Ile 47 |
|
7 |
4AAQ7 |
327.05 |
-9.99 |
Phe 97, Met -9.04199, Ile 202,-10.11 Met 103, Ile 21-9.215, Pro 193, He 149, Tyr 158, Ala 157, Met 161 |
|
8 |
PTQ8 |
1.05 |
-9.04 |
Phe 149, Pro 193, Gly 192, Met 199, Met 161, Ile 215, Phe 145, Leu 218, Tyr 158, Met 103 |
|
9 |
2APQ9 |
183.42 |
-10.11 |
Pro 193, Phe 149, Met 199, Ile 202, Ile 215, Leu 207, Leu 218, Gln 214, Ala 157, Met 155, Thr 158, Ala 157, Met 103 |
|
10 |
PABAQ10 |
353.45 |
-9.21 |
Gln 66, Asp 64, Phe 41, Leu 63, Gly 40, Ile 95, Val 65, Phe 97, Ile 122, Gly 96, Ile 95 |
|
11 |
Standard (Isoniazid) |
78.99 |
-5.89 |
Ile 95, Thr 39, Leu 63, Phe 41 |
|
Sl.No |
Compound code |
Physicochemical Properties |
|||
|---|---|---|---|---|---|
|
MW |
RB |
TPSA (Å) |
ESOL Class |
||
|
1 |
PNAQ1 |
342.36 |
4 |
83.63 |
M.Sol |
|
2 |
SMOQ2 |
457.51 |
6 |
118.39 |
M.Sol |
|
3 |
SNAQ3 |
376.44 |
4 |
106.35 |
M.Sol |
|
4 |
SGQ4 |
432.51 |
5 |
144.73 |
M.Sol |
|
5 |
DMUQ5 |
359.39 |
3 |
81.81 |
M.Sol |
|
6 |
AUQ6 |
331.33 |
3 |
103.53 |
Sol |
|
7 |
4AAQ7 |
339.40 |
4 |
54.88 |
M.Sol |
|
8 |
PTQ8 |
311.39 |
3 |
37.81 |
M.Sol |
|
9 |
APQ9 |
298.35 |
3 |
50.7 |
M.Sol |
|
10 |
PABAQ10 |
341.37 |
4 |
75.11 |
M.Sol |
|
Sl.No. |
Compound Code |
Pharmacokinetics Properties |
|||||
|---|---|---|---|---|---|---|---|
|
GIA |
BBB P |
Pgp S |
CYP1 |
CYP2 |
Log Kp (cm/s) |
||
|
1 |
PNAQ1 |
High |
No |
No |
Yes |
Yes |
-4.69 |
|
2 |
SMOQ2 |
Low |
No |
No |
No |
Yes |
-5.7 |
|
3 |
SNAQ3 |
High |
No |
No |
Yes |
Yes |
-5.79 |
|
4 |
SGQ4 |
Low |
No |
No |
Yes |
Yes |
-6.41 |
|
5 |
DMUQ5 |
High |
No |
No |
Yes |
No |
-6.53 |
|
6 |
AUQ6 |
High |
No |
No |
No |
No |
-6.62 |
|
7 |
4AAQ7 |
High |
Yes |
No |
Yes |
Yes |
-4.77 |
|
8 |
PTQ8 |
High |
Yes |
Yes |
Yes |
Yes |
-4.12 |
|
9 |
APQ9 |
High |
Yes |
No |
Yes |
Yes |
-4.82 |
|
10 |
PABAQ10 |
High |
No |
No |
Yes |
Yes |
-4.9 |
|
Sl.No. |
Compound Code |
Lipophilicity |
||||
|---|---|---|---|---|---|---|
|
I Log P |
X Log P |
W Log P |
M Log P |
Cons. Log P |
||
|
1 |
PNAQ1 |
2.89 |
5.21 |
5.47 |
3.52 |
3.89 |
|
2 |
SMOQ2 |
2.83 |
4.78 |
6.03 |
2.43 |
3.83 |
|
3 |
SNAQ3 |
2.51 |
3.95 |
4.77 |
2.23 |
3.14 |
|
4 |
SGQ4 |
2.07 |
3.44 |
4.08 |
2.43 |
2.73 |
|
5 |
DMUQ5 |
3.26 |
2.76 |
2.44 |
2.25 |
2.59 |
|
6 |
AUQ6 |
2.2 |
2.39 |
2.42 |
1.79 |
2.42 |
|
7 |
4AAQ7 |
3.17 |
5.07 |
5.24 |
3.58 |
4.33 |
|
8 |
PTQ8 |
3.42 |
5.75 |
5.35 |
4.28 |
4.7 |
|
9 |
APQ9 |
2.98 |
4.65 |
4.44 |
2.98 |
3.74 |
|
10 |
PABAQ10 |
2.76 |
4.91 |
4.74 |
2.23 |
3.65 |
Cons. Log P- Consensus Log P
|
Sl.No. |
Compound Code |
Drug Likeness |
|||||
|---|---|---|---|---|---|---|---|
|
No. of Violations |
BAS |
||||||
|
L |
G |
V |
E |
M |
|||
|
1 |
PNAQ1 |
0 |
0 |
0 |
0 |
1 |
0.55 |
|
2 |
SMOQ2 |
0 |
1 |
0 |
1 |
0 |
0.55 |
|
3 |
SNAQ3 |
0 |
0 |
0 |
0 |
0 |
0.55 |
|
4 |
SGQ4 |
0 |
0 |
1 |
1 |
0 |
0.55 |
|
5 |
DMUQ5 |
0 |
0 |
0 |
0 |
0 |
0.55 |
|
6 |
AUQ6 |
0 |
0 |
0 |
0 |
0 |
0.55 |
|
7 |
4AAQ7 |
0 |
0 |
0 |
0 |
1 |
0.55 |
|
8 |
PTQ8 |
1 |
0 |
0 |
0 |
1 |
0.55 |
|
9 |
APQ9 |
0 |
0 |
0 |
0 |
0 |
0.55 |
|
10 |
PABAQ10 |
0 |
0 |
0 |
0 |
0 |
0.56 |
L-Lipinski, G-Ghose, V-Veber, E-Egan, M-Muegge rule, BAS- Bioavailability Score
|
Sl.No. |
Sample code |
H37Rv stain (sensitive Strain) |
I2487 Stain (Resistant Strain) |
||||||
|---|---|---|---|---|---|---|---|---|---|
|
250 |
500 |
1000 |
1500 |
250 |
500 |
1000 |
1500 |
||
|
1 |
SMOQ2 |
S |
S |
S |
S |
S |
S |
S |
S |
|
2 |
SNAQ3 |
R |
R |
S |
S |
R |
R |
S |
S |
|
3 |
4AAQ7 |
S |
S |
S |
S |
S |
S |
S |
S |
|
4 |
2APQ9 |
R |
R |
S |
S |
R |
R |
R |
S |
|
5 |
PABAQ10 |
R |
R |
S |
S |
R |
R |
R |
S |
S: Sensitive R:Resistant
Pharmacokinetic Prediction
ADME screening was checked to predict the various physiochemical and pharmacokinetic properties of the designed 4-anilino quinazoline derivatives and also to predict their drug-likeness properties. The Lipinski's rule of five is a rule, to evaluate the drug-likeness of a molecule or compound. The rule states that an orally active drug can be less than 5 hydrogen bond donors, less than 10 hydrogen bond acceptors, MW should be less than 500 daltons, and the octanol-water partition coefficient (log P) does not exceed 5. Based on these calculations, the designed ligands satisfies the Lipinski's rule of five properties. The results were given in Table 6; Table 5; Table 4; Table 3.
Anti-tubercular Activity
Compound SMOQ2 and 4AAQ7 showed sensitivity in both H37Rv (Sensitive stain) and I2487 (Resistant strain) at the concentration of 250, 500, 1000 and 1500 mcg/mL. (Table 7).
Conclusion
In the present study, a series of novel 4-anilino Quinazoline derivatives were designed and docked into the active site of the protein Enoyl-Acyl carrier protein Reductase Mycobacterium tuberculosis (InhA) (PDB ID: 4TZK) as an anti-tubercular target. Molecular docking study showed that compound SMOQ2 (-9.66 kcal/mol), SNAQ3 (-9.31 Kcal/mol), 2APQ9 (-9.19 kcal/mol), 4AAQ7 (-8.85 Kcal/mol) and PABAQ10 (-8.8 kcal/mol) possess the highest binding affinity towards the target InhA compared to the standard drug Isoniazid (-5.6 Kcal/mol). The results of anti-tubercular activity revealed that quinazoline compounds containing oxazole ring and acetophenone group exhibited significant activity.