Syntheses and identification of cefotaxime-non-transition metal complexes
Abstract
The coordination chemistry of the biologically active cefotaxime sodium and, in particular, the mode of its interaction with some metal ions of electronic configuration d0 (alkaline earth) and others, Zn(II), Pb(II), and Ce ions with the electronic configuration d 10 has been investigated. Seven complexes were synthesized, isolated in the solid-state, and characterized by elemental analyses, conductivity measurements, IR and UV/VIS spectra, as well as thermal analyses. Based on the obtained experimental data and literature, the structural formulae to these complexes were suggested and formulated as [Mg(cef)2].2H2O (1), [Ca(cef)2].2H2O (2) [Sr(cef)2].2H2O (3), [Ba(cef)2].2H2O (4), [Zn(cef)2(H2O)2] (5), [Pb(cef)2(H2O)2].4H2O (6) and [Ce(cef)2(H2O)2].3H2O (7). The data obtained show that cefotaxime interacted with metal in a molar ratio of 2:1, respectively. Cefotaxime bonded to metal ions in the anionic form as a bidentate ligand through the lactam carbonyl (C=O) and the carboxylate group (COO-). Tetrahedral and octahedral shapes were proposed as the most likely geometries associated with a metal having such electronic configurations. The absorption bands observed in the electronic spectrum of free cefotaxime are also observed with some shifts in the spectra of its complexes, indicating their formation. The absorption bands of free cefotaxime and its complexes were assigned to electronic transitions. The thermal analyses date strongly support the structures proposed for the complexes and indicate the formation of the corresponding metal oxide as a final decomposition product.
Keywords
Synthesis, characterization, non-transition, metal complexes, cefotaxime
Introduction
The organic drug named cefotaxime is well known and its structure is elucidated in Figure 1. This compound contains many different active sites and used as an antibiotic from the third generation of cephalosporins. The cefotaxime (cef) was used to treat a wide range of bacterial inferctions and prevent infections during certain surgeries, as well as stopping bacterial growth. The most common mechanism proposed for cephalosporins resistance has been studied (Bergan, 1987; Neu, 1987; Ramotowska, Wysocka, Brzeski, Chylewska, & Makowski, 2019). These series have variable susceptibility to beta-lactamase produced by gram-negative than the first generation of cephalosporins (Zakaria, Afifi, & Elkhodairy, 2016).
The toxicological and pharmacological properties of many drugs are very probably modified when they are connected to metal ions in the form of complexes (Sorenson, 1990). Very few articles dealt with the coordination chemistry and the biological activity of cephalosporins with copper, which has proved favorable in diseases like tuberculosis, gastric ulcers, rheumatoid arthritis, and cancers (Brown, Smith, Teape, & Lewis, 1980; Sorenson, 1976; Williams, 1971). Few authors studied the coordination and biological activity of some cefotaxime-transition metal complexes (M = Mn(II), Fe(III), Co(II), Ni(II), Cu(II) and Cd(II)) in different ratios (Anacona & Estacio, 2006; Anacona & Rodriguez, 2005; Kondaiah, Chowdary, Reddy, & Rao, 2017). The solid complexes were isolated and identified with the usual techniques and screened for anti-bacterial activities. The results are compared with the activity of cefotaxime (Anacona & Silva, 2005; Faraj & Salih, 2020; Reiss, Chifiriuc, Amzoiu, & Spînu, 2014).
In this article, seven complexes are obtained during the reaction of cefotaxime with some non-transition metal ions with electronic configurations d 0 and d 10. The solid complexes were isolated in the solid-state and the identified and their chemical properties were characterized through elemental, molar conductivity measurements, vibrational and electronic spectra, as well as thermal analyses.
Materials and Methods
All chemicals used for the preparation of the complexes were of analytical reagent grade, commercially available from Sigma-Aldrich Chemical Co and used without further purification. These chemicals are Cefotaxime sodium, MgCl2·6H2O, CaCl2·2H2O, SrCl2·6H2O, BaCl2·2H2O, ZnBr2, Pb(CH3COO)2·3H2O, Ce(SO4)2 .
Synthesis
The cefotaxime−-M2+ complexes were prepared by mixing sodium cefotaxime (2.0 mmol) and the corresponding metal salts (1.0 mmol) in EtOH (50.0 mL). The reaction mixture was then stirred at room temperature for ca. 8 h, then left to stand overnight. The volume was reduced under vacuum. The precipitate was filtered off, washed with H2O and MeOH and then dried under vacuum at room temperature.
[Mg(cef)2].2H2O (1): C32H36MgN10O16S4, (969.25); yellow, Λm = 4. Anal. C, 39.12 (39.66); H, 3.80 (3.74); Mg, 2.84 (2.51); N, 14.73 (14.45); S,13.44 (13.23).
[Ca(cef)2].2H2O (2): C32H36CaN10O16S4 (985.02); yellow, Λm = 5. Anal. C, 38.94 (39.02); H, 3.72 (3.68); Ca, 4.32 (4.07); N, 13.96 (14.22); S, 13.24 (13.02).
[Sr(cef)2].2H2O (3): C32H36N10O16S4Sr, (1032.57); yellow, Λm = 3. Anal. C, 37.52 (37.22); H, 4.86 (3.51); N, 13.24 (13.56); S, 12.50 (12.42); Sr, 8.92 (8.49).
[Ba(cef)2].2H2O (4): C32H36BaN10O16S4, (1082.27); yellow, Λm = 7. Anal. C, 53.32 (35.51), H, 3.27 (3.35); Ba, 12.23 (12.69); N, 12.65 (12.94); S, 11.89 (11.85).
[Zn(cef)2(H2O)2] (5): C32H36N10O16S4Zn, (1010.34); yellow, Λm = 9. Anal. C, 37.86 (38.04), H, 3.84 (3.59), N, 13.12 (13.86); S, (12.59) 12.69; Zn, 6.05 (6.47).
[Pb(cef)2(H2O)2].4H2O (6): C32H44N10O20PbS4, (1224.21); yellow, Λm = 5. Anal. C, 30.87 (31.40); H, 5.52 (3.62); N, 11.97 (11.44); Pb, 15.86 (16.93); S, 10.52 (10.48).
[Ce(cef)2(H2O)2].3H2O (7): C32H42CeN10O19S4 (1139.11); yellow, Λm = 6. Anal. C, 33.41 (33.74); H, 3.17 (3.72); Ce, 12.25 (12.30); N, 12.20 (12.30); S, 11.21 (11.26).
(calculated values in parentheses and molar conductivity Λm in Ω-1cm2 mol-1).
Instrumentation and physical measurements
Elemental analyses (C, H, N, S) of the dried and pure samples were achieved on an automatic CHNS Vario EL III Germany elemental analyzer (Microanalysis Department of Cairo University, Cairo, Egypt). Metal contents were gravimetrically determined as metal oxides or by atomic absorption method using a spectrometer model PYE-UNICAM SP 1900 fitted with the corresponding lamp. The counter ions associated with the metals in their used salts: chloride, bromide, acetate and sulphate, were tested in the prepared complexes using the usual known procedures (Jeffery, Bassett, Mendham, & Denney, 1989). Infrared spectra were recorded on FT IR 640 PLUS using KBr disks in the range 4000−400 cm- 1. Thermogravimetric analyses were achieved under N2 atmosphere using TGA-50H Shimadzu with a heating rate of 10 oC min−1. Electronic spectra of the free ligand, cefotaxime and its metal complexes were carried out for solutions in DMSO using UV-3101PC Shimadzu Spectrophotometer in the range 200–800 nm. Molar conductivity measurements (1´10−3 M solutions, DMSO) of the cef-M complexes were carried out at room temperature using Jenway 4510 conductivity meter.
Results and Discussion
Cefotaxime non-transition metal complexes; [Mg(cef)2].2H2O, [Ca(cef)2].2H2O, [Sr(cef)2].2H2O, [Ba(cef)2].2H2O, [Zn(cef)2(H2O)2], [Pb(cef)2(H2O)2].4H2O, and [Ce(cef)2(H2O)2].3H2O were synthesized and isolated as stable solids at room temperature. The chemical formulas of these complexes were proposed based on their reaction ratios, elemental analyses, as well as their qualitative and thermal analyses. The conductivities measured in DMSO for the prepared complexes were ranging from 3.0 – 9.0 Ω−1cm−1mol−1, which indicate a non-electronic nature (Geary, 1971) and agree quite well with the proposed formulas.
|
cef |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
Assignment* |
|---|---|---|---|---|---|---|---|---|
|
3431 br |
3412 br |
3398 br |
3462 br |
3412 br |
3410 br |
3348 br |
3352 br |
ν(O−H), H2O |
|
3350 m |
3352 sh |
3373 sh |
3381 w |
3349 sh |
3361 sh |
3382 w |
3312 br |
ν(N-H), NH2 |
|
3259 w |
3248 sh |
3261 sh |
3253 sh |
3265 sh |
3268 sh |
3253 w |
3243 sh |
|
|
3086 w |
3049 sh |
3024 sh |
3074 sh |
3076 sh |
3053 sh |
3063 w |
3035 w |
ν(C-H), aromatic |
|
2938 w |
2941 w |
2934 w |
2963 w |
2945 w |
2945 w |
2954 w |
2923 w |
ν(C-H), aliphatic |
|
2822 w |
2820 w |
2832 w |
2837 w |
2832 sh |
2812 sh |
2856 w |
2817 w |
|
|
1761 vs |
1773 s |
1767 vs |
1768 s |
1771 s |
1774 s |
1769 s |
1770 s |
ν(C=O), lactam |
|
1647 vs |
1655 vs |
1658 sh |
1653 vs |
1649 sh |
1652 s |
1654 vs |
1662 sh |
ν(C=O), ester; amide |
|
1583 s |
1626 vs |
1649 vs |
1637 vs |
1624 vs |
1632 s |
1615 vs |
1622 vs |
νas(C=O), (COO-) |
|
1538 s |
1542 s |
1535 s |
1540 s |
1543 s |
1542 s |
1552 s |
1552 s |
(NH2) |
|
1405 s |
1386 s |
1394 s |
1396 s |
1398 s |
1374 s |
1397 s |
1389 s |
νs(C=O), (COO-) |
|
1243m |
1235 s |
1237 s |
1246 s |
1235 s |
1243 s |
1260 s |
1234 m |
νs(C–N) |
|
- |
562 w |
557 w |
548 w |
549 w |
556 w |
564 w |
568 w |
νs(M–O) |
|
- |
503 w |
496 w |
483 w |
493 w |
495 w |
485 w |
480 w |
* v, very;s, strong; m, medium; w, weak; br, broad; and sh, shoulder.
|
cef |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|
|---|---|---|---|---|---|---|---|---|
|
νas(C=O) |
1583 |
1626 |
1649 |
1637 |
1624 |
1632 |
1615 |
1622 |
|
νas(C=O) |
1583 |
1626 |
1649 |
1637 |
1624 |
1632 |
1615 |
1622 |
|
ν(COO)- |
178 |
240 |
253 |
241 |
226 |
258 |
218 |
233 |
|
Transition |
Cef |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
|---|---|---|---|---|---|---|---|---|
|
|
275 |
283 |
285 |
278 |
282 |
289 |
288 |
284 |
|
|
320 |
322 |
319 |
321 |
328 |
325 |
322 |
326 |
|
|
386 |
398 |
403 |
405 |
396 |
403 |
398 |
403 |
|
Compound |
Decomp. |
Tmax (OC) |
Lost species |
% weight loss |
|
|---|---|---|---|---|---|
|
Step |
Found |
Cal. |
|||
|
Cefotaxime |
1st step |
195-400 |
C16H16N5O5S2 |
89.02 |
88.48 |
|
C16H16N5NaO7S2 |
Total loss |
89.02 |
88.48 |
||
|
Residue |
NaO2 |
10.98 |
11.51 |
||
|
[Ca(cef)2].2H2O |
1st step |
95 |
2H20 |
3.94 |
3.65 |
|
C32H36N10O16S4Ca |
2nd step |
190-370 |
C32H32N10O13S4 |
89.63 |
90.64 |
|
Total loss |
93.57 |
94.29 |
|||
|
Residue |
Ca0 |
6.43 |
5.76 |
||
|
[Sr(cef)2].2H2O |
1st step |
100 |
2H20 |
3.57 |
3.48 |
|
C32H36N10O16S4Sr |
2nd step |
300-430 |
C32H32N10O13S4 |
85.84 |
86.47 |
|
Total loss |
89.41 |
89.95 |
|||
|
Residue |
SrO |
10.59 |
10.04 |
||
|
[Ba(cef)2].2H2O |
1st step |
94 |
2H20 |
3.32 |
3.32 |
|
C32H36N10O16S4Ba |
2nd step |
300-450 |
C32H32N10O13S4 |
82.69 |
82.42 |
|
Total loss |
85.92 |
85.74 |
|||
|
Residue |
BaO |
14.08 |
14.15 |
||
|
[Zn(cef)2(H2O)2] |
1st step |
163 |
2H20 |
3.94 |
3.56 |
|
C32H36N10O16S4Zn |
2nd step |
347 |
C32H32N10O13S4 |
87.69 |
88.37 |
|
Total loss |
91.63 |
91.93 |
|||
|
Residue |
ZnO |
8.37 |
6.47 |
||
|
[Pb(cef)2(H2O)2].4H2O |
1st step |
95 |
2H20 |
3.01 |
2.94 |
|
C32H44N10O20S4Pb |
2nd step |
154 |
4H2O |
5.94 |
5.88 |
|
3rd step |
230-380 |
C32H32N10O13S4 |
73.65 |
72.93 |
|
|
Total loss |
82.60 |
81.75 |
|||
|
Residue |
PbO |
17.40 |
18.23 |
||
|
[Ce(cef)2(H2O)2].3H2O |
1st step |
93 |
2H20 |
3.26 |
3.16 |
|
C32H42N10O19S4Ce |
2nd step |
160 |
4H20 |
5.03 |
4.74 |
|
3rd step |
290-480 |
C32H32N10O13S4 |
75.84 |
77.09 |
|
|
Total loss |
84.13 |
84.99 |
|||
|
Residue |
CeO2 |
15.87 |
15.01 |
||
Vibrational spectra
The IR spectral data of the pure cefotaxime and M-cef complexes and the assignments of the defined characteristic bands are summarized in Table 1. The spectra of the free cef and its metal complexes, M-cef, reveal approximately the same absorption pattern in the region of 3500–2800 cm- 1. The expected bands in this region may be due to ν(O-H), ν(N-H), ν(C-H) aromatic, and ν(C-H) aliphatic vibrations (Guzler & Germlich, 2002). All these bands are observed in the spectra of the free cefotaxime and its metal complexes in the typical regions with some shifts in the band frequencies. This shift can be attributed to the changes in the electronic density distribution among aromatic rings and the attached functional groups because of the proposed chelation in the metal complexes.
The carbonyl stretching vibrations of lactam ν(C=O) is observed in the spectrum of the free cefotaxime at 1761 cm-1 as a very strong band (Anacona et al., 2005; Anacona et al., 2005). The corresponding vibrations in the spectra of cefotaxime complexes were observed at somewhat higher values in the range 1767–1774 cm-1. This behavior is taken as evidence for the engagement of the lactam carbonyl in coordination with metal ions. The ring carbonyl absorption frequency will be shifted to higher wave numbers as the ring becomes more and more strained (Anacona & Osorio, 2008; Anacona, 2001; Singh, Grover, Kumar, & Jain, 2010). The amide carbonyl is observed at 1647 cm- 1 overlapped with the ester carbonyl stretching vibration (Anacona et al., 2005; Singh et al., 2010). The corresponding motions in metal complexes are observed at almost the same frequency in the region 1658–1649 cm- 1. This suggests that none of these groups are involved in binding with metal ions. The characteristic bending motion of the amide group (NH2) in the free cefotaxime spectrum and in the spectra of its complexes approximately occurs in the same region 1535 – 1552 cm- 1, also suggesting the coordinating nature of this group in our complexes (Bellamy, 1975; Guzler et al., 2002; Nakamoto, 1997).
The carboxylate group exhibits several modes for binding metal ions (Nakamoto, 1997). For a monodentate mode of the carboxylate group, the antisymmetric stretch, νas(COO), will increase and the symmetric stretch, νs(COO), will decrease as the metal-oxygen bond become stronger (more covalent character) in the complexes. The νas(COO) motion of the carboxylate group in the spectrum of cefotaxime is observed at 1583 cm- 1 as a strong band while the corresponding motion in the spectra of the complexes lies at a higher frequency in the range 1622–1649 cm- 1. The symmetric motion of the carboxylate group, νs(COO), is assigned at 1405 cm- 1 in the spectrum of cefotaxime. The complexes spectra exhibit this band at lower frequencies region of 1374–1398 cm- 1, which is the typical behavior for a monodentate carboxylate group (Guzler et al., 2002; Nakamoto, 1997). Furthermore, the separation between the asymmetric and symmetric stretching motions of the carboxylate group in the complexes spectra are given in Table 2 and is shown to be > 200 cm- 1, which is a further indication for the monodentate nature of the carboxylate group in these complexes (Nakamoto, 1997).
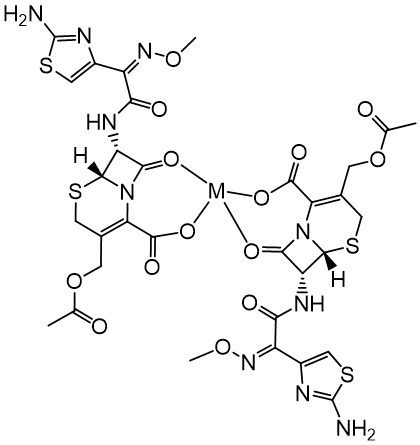
Finally, the complexes spectra reveal bands in the regions 562–548 cm- 1 and 503– 480 cm- 1 may be due to the stretching motions corresponding to ν(M-O) (Anacona, 2001; Nakamoto, 1997; Shungu, Weinberg, & Gadebusch, 1983). These bands are not observed in the spectrum of the free cefotaxime and significantly support the proposed mode of chelation as a nonnegatively charged anion with a bidentate nature through the lactam carbonyl and the carboxylate groups. The most probable geometry associated with the metals of d0 and d10 configurations is likely to be tetrahedral and octahedral, as shown in Figure 4 and Figure 5, respectively.
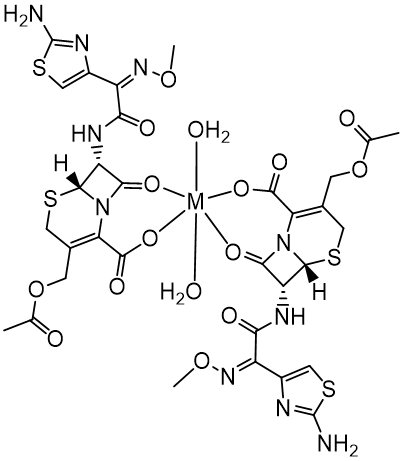
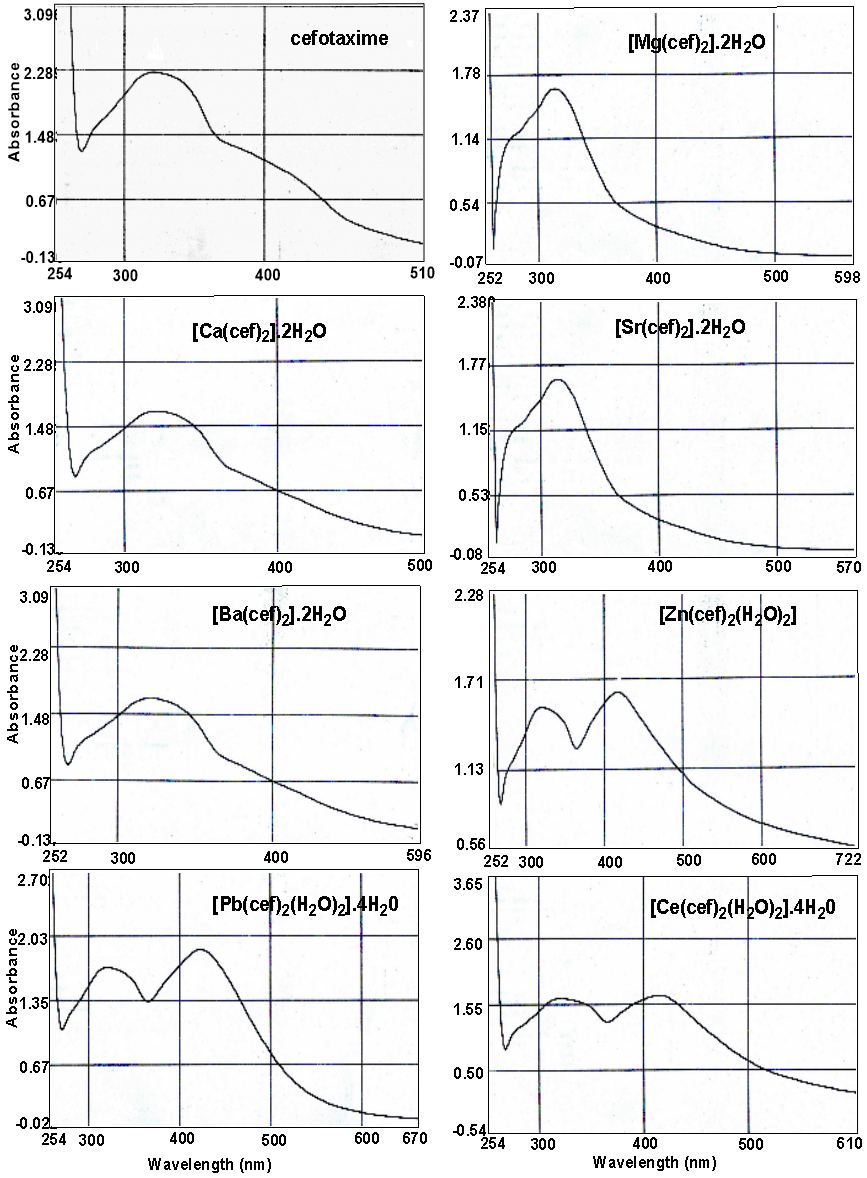
Electronic spectra
The ultraviolet/visible spectra of the free base cefotaxime and its complexes in dimethylsulfoxide and given in Figure 2. The assignments of absorption bands into electronic transitions are given in Table 3—two clear observation emerging on comparing the spectra of free cefotaxime with its complexes. The first one is that the spectra reveal the same absorption pattern with some expected shifts attributed due to the complexation interaction between cefotaxime and metal ions. The second observation is the absence of any absorption may be attributed to the d – d transitions in the spectra of metal complexes. Such observation is expected for d 0 and d 10 complexes. Three absorption peaks are observed in the spectra, which may be attributed due to the electronic transition inside the cefotaxime base. The first one is observed in the rang 275–289 nm and assigned to the transition due to molecular orbital energy levels originating in the N–C– N moiety (Franchini, Giusti, Preti, Tassi, & Zannini, 1985; Hadjikostas, Katsoulos, & Shakhatreh, 1987). The second band observed in the region 320–328 nm is related to the transitions within the triazole and 1,3-triazole moieties (Jasim, Said, & Ali, 2017). The third band in 38–405 nm regions is attributed to intra-ligand transitions of the type, which is in accordance with the literature data for transitions due to sulfur atoms (Franchini et al., 1985; Hadjikostas et al., 1987). This fact proves that sulfur atoms are not involved in the coordination to metal ions.
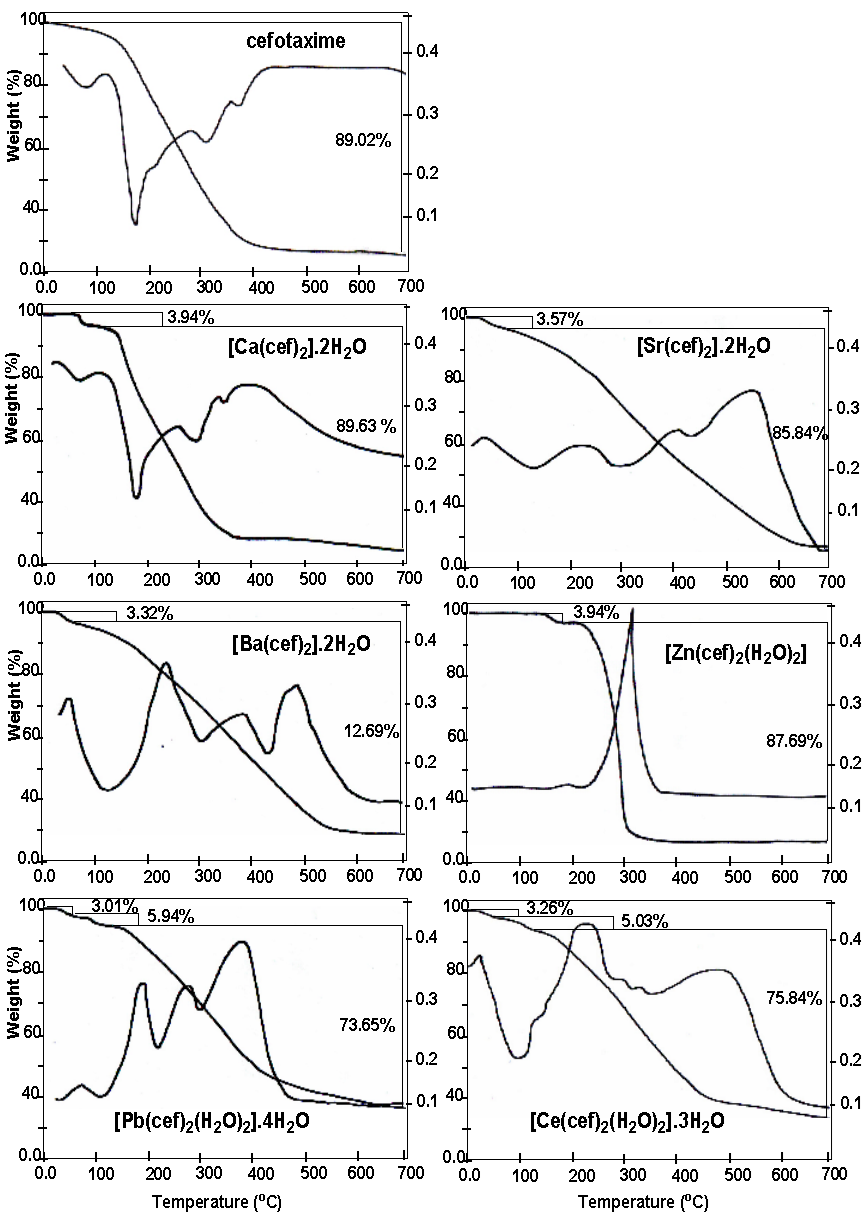
Thermal degradation
Thermal degradation of cefotaxime and its metal complexes were carried out under nitrogen atmosphere using thermogravimetric (TGA) and differential thermogravimetric (DTG) analyses. The obtained thermograms are given in Figure 3, and the maximum temperature values for decomposition, along with the corresponding weight loss values, are given in Table 4.
The data obtained from the mode of free cefotaxime decomposition reveal that the organic moiety is lost at a wide range of temperature distributed among several maxima in the range 195 – 400oC. The weight loss associated with this process is 89.02%, consistent with the calculated 88.48%. The residue, which is proposed to be NaO2, gives a theoretical weight loss of 11.51%, agrees quit well with the found value of 10.98%. Such mode of decomposition for free cefotaxime was also observed in the thermograms of its metal complexes but at temperature range shifted to higher values. This gives clear evidence on the formation and structure of the complexes.
The alkaline earth complexes [Ca(cef)2].2H2O, [Sr(cef)2].2H2O, and [Ba(cef)2].2H2O are nearly decomposed in a similar fashion reflecting the similar chemical formula proposed for the complexes. The first step of thermal degradation is observed in the temperature range 94–100oC and associated with a weight loss of 3.94%, 3.57%, 3.32%, corresponding to the loss of two lattice water molecules in the complexes [Ca(cef)2].2H2O, [Sr(cef)2].2H2O, and [Ba(cef)2].2H2O, respectively. These values consistent very will the calculated values of 3.65%, 3.485, and 3.32% for the three complexes, respectively. The corresponding metal oxide (MO) was proposed to be the final decomposition product because it gives found values of 6.43%, 10.59%, and 14.08% in consistence with the calculated values of 5.76%, 10.04% and 14.155, respectively.
The thermogram of the complex [Zn(cef)2(H2O)2] shows two degradation steps. The first one lies at a relatively higher temperature, 163oC and may be attributed due to the loss of the two coordinating water molecules. The theoretical weight loss associated with this step is 3.56 % agrees quite well with the found value of 3.94%. The second step is the loss of the organic moiety at a temperature maximum of 347oC, with a match in the calculated and practical values. The metallic residue for such decomposition reaction is ZnO for the consistency in found and calculated values of 8.37% and 6.47%, respectively.
The other two complexes, Pb(cef)2(H2O)2].4H2O and [Ce(cef)2(H2O)2].3H2O, reveal very similar decomposition reactions reflecting the similarity in the chemical composition. The two complexes show three degradation stages; the first one is observed at the lower range (90–100oC) of temperature characteristic for lattice water. The found weight loss associated with this step matches very well the calculated one and correspond to the loss of two uncoordinated water molecules, see Table 4. The second degradation step lies in the range of 150–160oC, corresponds to the loss of four and three coordinated water molecules in the complexes Pb(cef)2(H2O)2].4H2O and [Ce(cef)2(H2O)2].3H2O, respectively. The found and calculate values of the weight loss in such step are in good agreement. The last step in this rection occurs in the temperature range 230– 480oC, corresponding to the loss of the organic moiety. The residue of 17.40% and 15.87% are consistent very well with the calculated values of 18.23% and 15.01% for PbO and CeO2 as a final decomposition product in the two complexes, respectively.
Conclusion
The formed complexes of cefotaxime with non-transition metal ions were synthesized and identified by microanalyses, conductivity measurements, infrared and UV-Vis spectra, besides thermal analyses. Based on the obtained experimental data and literature indicate, the structural formulae to these complexes were suggested and formulated as [Mg(cef)2].2H2O (1), [Ca(cef)2].2H2O (2) [Sr(cef)2].2H2O (3), [Ba(cef)2].2H2O (4), [Zn(cef)2(H2O)2] (5), [Pb(cef)2(H2O)2].4H2O (6) and [Ce(cef)2(H2O)2].3 H2O (7). According to the IR data of these non-transition metal ion complexes with cefotaxime, base ligand behaves in the prepared mononuclear complexes as a bi-dentate -OO ligand. The two bonding sites are the oxygen atom of the ionized carboxylate, -COO- and the O atom of the C=O in lactam carbonyl. Complexes 1-4 contain two lattice water molecules; however, complexes 5-7 also contained two coordinated water molecules. Thermal studies of the free cefotaxime and the prepared complexes showed cefotaxime have a similar coordination behavior in all complexes and decomposed at higher temperatures in comparison with the free ligand.
Conflict of Interest
The authors declare that they have no conflict of interest for this study.
Funding Support
The authors declare that they have no funding support for this study.